Болезнь Виллебранда — Википедия
Болезнь Виллебранда-Диана — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии.
Заболевание наследуется по принципу аутосомного доминирования. Возможно наследование и по аутосомно-рецессивному типу (2 и 3 тип болезни).
Распространенность болезни Виллебранда составляет 1 на 800—1000. В отдельных источниках указывается, что частота случаев равна 2%[3].
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда, который участвует в адгезии тромбоцитов на коллагене и защищает VIII фактор от протеолиза. При дефиците Фактора Виллебранда VIII фактор подвергается протеолизу, и его содержание в плазме снижается. Кроме того, при болезни Виллебранда снижается содержание серотонина и развивается патологическая дилатация сосудов и повышение их проницаемости. При болезни Виллебранда наблюдаются самые длинные кровотечения, т.к. у больных нарушены все три звена гемостаза.
- Ангиогемофилия;
- Атромбопеническая пурпура;
- Атромбоцитопеническая пурпура;
- Геморрагическая капилляропатия;
- Конституциональная тромбопатия, конституциональная тромбопатия фон Виллебранда-Юргенса;
- Наследственная псевдогемофилия; синдром Юргенса;
- Сосудистая гемофилия, псевдогемофилия.
Различают три типа болезни Виллебранда.
- 1-й тип обусловлен частичным количественным дефицитом фактора Виллебранда. При этом мультимерная структура его сохранена. Имеется снижение прокоагулянтной активности фактора VIII, агрегации тромбоцитов, индуцированной ристоцетином, ристоцетинкофакторной активности, антигена фактора Виллебранда. Частота данной формы составляет от 75 % до 80 % всех случаев болезни Виллебранда. Наследование аутосомно-доминантное.
- 2-й тип обусловлен качественными изменениями фактора Виллебранда, связанными с нарушением формирования мультимеров, и подразделяется на подтипы: 2A, 2B, 2M, 2N.
- Фенотип подтипа 2A является результатом нарушения двух различных механизмов: дефекта синтеза высокомолекулярных мультимеров и повышения протеолиза фактора Виллебранда. При подтипе 2B отмечается повышенное сродство фактора Виллебранда к рецептору на мембране тромбоцитов гликопротеину Ib.
- Подтип 2N характеризуется нормальным уровнем фактора Виллебранда и низкой прокоагулянтной активностью, что обусловлено нарушением связи фактора VIII и фактора Виллебранда.
Наследование болезни Виллебранда 2-го типа аутосомно-доминантное, за исключением подтипа 2N, где оно рецессивное. Частота встречаемости данных форм составляет от 5 % до 15 % всех случаев болезни Виллебранда.
- 3-й тип — наиболее тяжелая форма с полным дефицитом фактора Виллебранда. Эта форма характеризуется отсутствием фактора Виллебранда в плазме, тромбоцитах и сосудистой стенке. Уровень фактора VIII ниже 10 %. Наследование — аутосомно-рецессивное. Заболевание проявляется у гомозигот с одинаковыми дефектными аллелями или двойных гетерозигот с двумя различными дефектными аллелями. У пациентов с 3-м типом имеется вероятность появления аллоантител к фактору Виллебранда. Частота встречаемости заболевания 3-го типа болезни Виллебранда менее 5 %.
Кроме того, существует тромбоцитарный тип болезни Виллебранда, который обусловлен мутацией в гене тромбоцитарного рецептора гликопротеина Ib, вследствие которой повышается чувствительность данного рецептора к высокомолекулярным мультимерам фактора Виллебранда. Фенотип аналогичен подтипу 2B.
- Приобретенный синдром Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативными заболеваниями, обусловлен появлением ингибитора против фактора Виллебранда, а также качественными аномалиями фактора VIII в связи с адсорбцией высокомолекулярных мультимеров патологическими белками.
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из слизистых полости рта, носа, внутренних органов. Симптомы кровоточивости варьируют от умеренно выраженных до крайне тяжелых, протекают преимущественно по микроциркуляторному типу. У пациентов с резким дефицитом фактора VIII наблюдаются обильные и продолжительные кровотечения (носовые, десневые, маточные), также кровоизлияния в мышцы и суставы. Кроме того, могут возникать длительные кровотечения при травмах, удалении зубов, операциях.
В детском возрасте часто бывают кровотечения из слизистых оболочек полости рта, носовые кровотечения, синяки на коже. Более тяжелое течение геморрагического диатеза отмечается во время или вскоре после перенесенных инфекционных заболеваний. Наиболее вероятным пусковым механизмом кровотечения на фоне инфекции является нарушение проницаемости сосудов. Вследствие этого появляются самопроизвольные кровотечения диапедезного типа.
Гематомы — кровоизлияния в подкожную клетчатку и мышечные ткани наблюдаются преимущественно после травм у больных с тяжелыми формами заболевания.
При болезни Виллебранда геморрагический синдром проявляется не всегда, периоды обострения чередуются с периодами полного или почти полного отсутствия геморрагий. У некоторых пациентов болезнь Виллебранда может сочетаться с признаками мезенхимальной дисплазии: повышенной растяжимостью кожи, слабостью связок с повышенной подвижностью суставов, пролабированием створок клапанов сердца.
Аутосомный тип наследования обусловливает одинаковую частоту возникновения болезни Виллебранда у пациентов обоих полов. У женщин вследствие особенностей физиологического строения организма, связанных с репродуктивной функцией, наблюдается более частое проявление геморрагических симптомов. Около 65 % женщин с болезнью Виллебранда страдают
Желудочно-кишечные кровотечения у пациентов с болезнью Виллебранда не являются преобладающей формой кровоточивости. Они могут быть вызваны приемом препаратов, влияющих на агрегацию тромбоцитов (ацетилсалициловая кислота и другие нестероидные противовоспалительные средства). Кроме того, источниками кровотечений являются латентные язвы желудка и двенадцатиперстной кишки, также эрозивные гастриты, геморроидальные узлы.
У пациентов с болезнью Виллебранда могут быть длительные кровотечения при операциях
Гемартроз — наиболее редкое проявление болезни Виллебранда, характерное для заболевания 3-го типа. Острый гемартроз сопровождается болевым синдромом, обусловленным повышением внутрисуставного давления. Сустав увеличен в объеме, кожа над ним гиперемирована и горячая на ощупь. Если гемартроз возник после травмы, нужно исключить дополнительные повреждения (внутрисуставной перелом, отрыв мыщелка, ущемление тканей). Рецидивирующие гемартрозы вызывают хронический синовит. На стадии синовита синовиальная оболочка гипертрофируется и становится основным источником кровоизлияния в сустав. При остром синовите гемартрозы могут рецидивировать, несмотря на трансфузии фактора свертывания VIII, что обусловлено воспалительным процессом в синовиальной оболочке. При хроническом синовите болевой синдром может отсутствовать, поскольку разрушена капсула сустава.
В отличие от гемофилии, при болезни Виллебранда дальнейшего прогрессирования патологического процесса и развития деформирующего остеоартроза, как правило, не наблюдается.
Кровоизлияния в головной и спинной мозг и их оболочки при болезни Виллебранда возникают в связи с травмой. В отдельных случаях причиной таких кровоизлияний может быть гипертонический криз или прием препаратов, значительно нарушающих гемостатическую функцию тромбоцитов (ацетилсалициловая кислота, бутадион и др.).
Учитывая аутосомно-доминантный тип наследования, генетический риск для потомства составляет 50 % независимо от пола плода[4].
Лечение зависит от типа заболевания. Выделяют два основных способа. При первом используются препараты плазмы с высоким содержанием фактора Виллебранда или препараты фактора VIII. В легких случаях может быть достаточно одного введения, при тяжелых травмах и операциях препараты вводят дважды в сутки 2—3 дня. Второй подход к лечению применим для легких форм. Пациентам назначается десмопрессин. Существует опасность привыкания при использовании десмопрессина более 2-х суток.
Болезнь Виллебранда — причины, симптомы, диагностика, лечение
Современная медицина шагнула далеко вперёд, но даже это не спасает от генетических и врождённых патологий. Врачи до сих пор активно ведут исследования этих проблем, одна из них – это количественный и качественный дефицит плазменного фактора Виллебранда. Каковы основные причины развития и симптомы? Что необходимо предпринять для лечения и профилактики?
Что это такое
Болезнь Виллебранда — что это такое? Другое название – ангиогемофилия.
Заболевание является разновидностью геморрагического диатеза, который передаётся по наследству. Но может быть и приобретённым.
Патология обусловлена дефицитом или малой активностью плазменного компонента VIII-го фактора свёртываемости крови. Диагностируется у 1-2 человек из десяти тысяч. Занимает третье место после тромбоцитопатии и гемофилии.

Часто наблюдается совместно с другими заболеваниями:
- соединительной дисплазией;
- слабостью связок;
- гипермобильностью суставов;
- высокой растяжимостью кожи;
- пролапсом клапанов сердца.
Развивается у мужчин и у женщин, но у последних протекает в более тяжело.
При недуге спонтанно появляются подкожные петехии, экзимозы. Повышается склонность к кровотечениям и к избыточной потере кровяной жидкости при травмах и операциях.
Важно! Патология возникает постепенно и без ярких симптомов, обнаружить самостоятельно на начальных стадиях крайне сложно и часто она переходит в более тяжёлую форму.
Почему появляется

Причины делят на две группы – внешние и внутренние. Врождённую форму провоцирует мутация в гене Виллебранда, то есть передаётся от матери к ребёнку. Приобретённая патология развивается из-за этого же, но только на фоне внешнего воздействия, например,
- постоянные высокие физические нагрузки;
- стрессы, невроз;
- на фоне беременности;
- от перенесённых инфекций.
Норма при хорошей свёртываемости крови – 10 мг/л. Если уровень повышен или понижен, то со временем проявляется симптоматика.
Как развивается
При внешнем воздействии или же из-за наследственности VIII фактор свёртывания распадается, а кровеносные сосуды становятся шире и в итоге проницаемость их стенок увеличивается. Из-за этого у больного часто возникают кровотечения, которые локализуются в разных местах и с разной интенсивностью.

Страдает полностью система гемостаза, то есть нарушается процесс образования тромбов для прекращения кровотечений, потому что для этого необходима высокая активность фактора Виллебранда.
Классификация
Патологию делят на несколько типов.
- Классический тип — I стадия. Самая распространённая форма, протекает легко и без серьёзных осложнений. Уровень плазменного количества немного снижен.
- II стадия – наблюдается у 25-30 % больных. Анализ крови в норме, но активность VIII-го компонента снижена.
- Тяжёлая степень — III стадия. Проявляются выраженные симптомы, анализы выдают недостаток в крови необходимых элементов. Люди с болезнью в данной форме – большинство инвалиды.
- Псевдогемофилия или тромбоцитарный тип – так назвал эту стадию открыватель-учёный. Количество элемента, отвечающего за свёртываемость в норме, но нарушена связь с изменёнными рецепторами тромбоцитов.

Симптомы
Болезнь Виллебранда имеет основные симптомы:
- кровотечения при малейших травмах;
- подкожные гематомы;
- маточная кровопотеря;
- длительный менструальный цикл.
У каждой формы существуют отличительные черты проявления, I и II тип сопровождается:
- частое носовое кровотечение;
- постоянные подкожные гематомы;
- повышенное количество крови при и после операций, травм;
- длительные месячные у женщин.
III тип самый тяжёлый и похож на гемофилию:
- гематомы и подкожные кровоизлияния;
- болезненные ощущения в местах синяков;
- затруднённые движения и отеки;
- кровоизлияния в сосудах —гемартроз— в матке, в желудке, урологические, внутричерепные.
Часто возникают десенные кровотечения, примеси крови в кале и моче.

Чем опасна
Если фактор Виллебранда повышен, то могут возникнуть следующие осложнения:
- анемия;
- большая кровопотеря;
- тромбоз;
- маточные кровотечения;
- летальный исход при тяжёлой форме;
- проблемы при беременности — угроза выкидыша, отслойка плаценты, гестоз, роды с осложнениями.
Патология не является приговором при вынашивании, главное – это нужно тщательно спланировать беременность, пройти скрининги и постоянно быть под наблюдением врача.
Болезнь Виллебранда у детей проявляется в первый год жизни, если передаётся по наследственному фактору. Поэтому новорождённых сразу же обследуют.

Диагностика
Чтобы выявить болезнь Виллебранда для диагностики используют следующие методы:
При необходимости врач может назначить дополнительные обследования, это зависит от состояния больного.

Терапия
При лёгкой форме болезни лечение проводится при необходимости, например, ввиду скорой операции, при травмах или в целях профилактики.
Самая уязвимая категория пациентов – это беременные женщины. Терапия заключается в употреблении препаратов и частых переливаниях кровяных компонентов. Лишь такими способами можно избежать избыточной кровопотери при родах.
Для лечения детей применяют средства:
- криопреципитаты;
- свежезамороженную плазму;.
- гомеопатические препараты.
Постоянный прием медикаментов необходим людям с тяжёлой формой патологии. Для этого назначают лекарства, которые имеют кровоостанавливающее воздействие и, содержащие компонент необходимый для свёртываемости.

Женщинам же рекомендовано дополнительно гормональное лечение.
Иногда, в редких случаях, проводят хирургическую операцию для перевязки сосудов или удаления некоторой части, чтобы купировать кровоизлияние.
Важно! Избавиться от болезни навсегда нельзя, возможно только снятие симптомов и профилактика.
При патологии, протекающей в тяжёлой стадии, пациенту иногда присваивается инвалидность. Но чаще люди, с содержанием плазменного элемента ниже 5 % не доживают и до тридцати лет.
Профилактика
Для динамичного лечения необходимо придерживаться правил профилактики:
- активный и здоровый образ жизни;
- избегать травм и опасных видов спорта;
- беременность планировать с гинекологом;
- выполнять предписания врача и употреблять прописанные препараты;
- следить за массой тела;
- регулярно наблюдаться у доктора.
Несмотря на то, что патология мало распространена, однако при игнорировании симптомов и отсутствии лечения может обернуться инвалидностью и летальным исходом. Главное вовремя диагностировать недуг и начать терапию. Поэтому не стоит пренебрегать профилактическими осмотрами у врача.
Недуг в наследство: чем опасна болезнь Виллебранда?
Сегодня согласно официальной статистике в России гемофилией страдают около 8,5 тысяч пациентов, хотя когда -то эта патология считалась царской болезнью. Гемофилия- наследственное заболевание, обычно проявляющееся только у мужчин, в то время как женщины выступают в качестве носительниц гена. Конечно, самой известной носительницей гемофилии в истории стала королева Виктория. Предположительно, эта мутация произошла в её генотипе de novo, так как в семьях ее родителей не были зарегистрированы больные гемофилией. Наиболее распространенные формы заболевания — классическая гемофилия типа А и гемофилия типа В, при которых развивается недостаточность VIII тромбообразующего фактора или фактора плазмы IX (Кристмаса).
В отличие от гемофилии, которой страдают только мужчины, существует еще один наследственный геморрагический диатез, которому подвержены и женщины — болезнь Виллебранда. Эта патология, наследуемая по аутосомно-доминантному типу, намного тяжелее диагностируется и зачастую пациент узнает о ней лишь во время тяжелых травм, хирургических вмешательств и других критических состояниях. В мире болезнь Виллебранда встречается с частотой у 1 человека из 100. В России по официальным данным распространенность составляет 0,001 %, однако многие эксперты полагают, что реальное число пациентов гораздо больше. Низкую выявляемость болезни можно объяснить преобладанием легких и бессимптомных форм, а также сложностью диагностики. Основная масса больных гемофилией и болезнью Виллебранда – люди трудоспособного возраста, именно поэтому на первый план выступают мероприятия, направленные на профилактику осложнений. При болезни Виллебранда возникает недостаточность фактора Виллебранда, белка, играющего важную роль в регуляции адгезии тромбоцитов к коллагену субэндотелия и защищающего VIII фактор от протеолиза. Кроме того снижается содержание серотонина и развивается патологическая дилатация сосудов и повышение их проницаемости. Стоит подчеркнуть, что именно при данной патологии наблюдаются самые длительные кровотечения, так как повреждены все три звена гемостаза. Существует также приобретенный синдром Виллебранда, характерный для пациентов с аутоиммунными и лимфопролиферативными заболеваниями и обусловленный образованием ингибитора против фактора Виллебранда.
Клиническое течение болезни Виллебранда может существенно снизить качество жизни, приводить к ранней и стойкой утрате трудоспособности, а в тяжелых случаях даже к летальному исходу. Долгое время заболевание рассматривалось как одна из форм гемофилии, так как многие проявления болезни Виллебранда схожи с симптомами данной нозологии. Однако лечение гемофилии не всегда оказывалось эффективным в случае болезни Виллебранда. В связи с этим в марте 2015 г вступил в силу новый Перечень лекарственных препаратов, в котором лечение болезни Виллебранда было вынесено в отдельный пункт.
К наиболее патогномоничным симптомам болезни Виллебранда относят кровотечения из слизистых полости рта, носа, внутренних органов, а также появление долго не проходящих синяков. В отличие от гемофилии, которую обнаруживают еще при рождение, болезнь Виллебранда, особенно ее легкие формы, диагностируют лишь в подростковом возрасте или при проведение инвазивных вмешательств, когда кровотечение бывает трудно остановить. Выраженность проявлений может варьировать от легких форм до весьма тяжелых вариантов с длительными кровотечениями самой разнообразной локализации, формированием гематом и кровоизлияний в мягкие ткани, суставы и внутренние органы. При таких формах дети уже к 12-15 годам становились инвалидами и редко доживали до 30 лет.
Сегодня благодаря достижениям мировой науки пациенты с нарушениями свертываемости крови, могут жить полноценной жизнью, получать образование, работать, успешно социализироваться. Продолжительность и качество их жизни находится на том же уровне, что и у здорового человека. При подозрение на болезнь Виллебранда необходимо исследование крови на качественные и количественных характеристики фактора Виллебранда. Наиболее часто применяются следующие диагностические тесты: ристомицин кофакторная активность, агрегация тромбоцитов с ристомицином, коллаген связывающая активность фактора Виллебранда, фактор VIII связывающая активность фактора Виллебранда или анализ мультимеров. Вместе с тем остается еще несколько вопросов, над которыми стоит работать. Как часто нужно проводить диагностику? Какие пациенты нуждаются в ней ? Как оценивать норму? Более того, активность фактора Виллебранда постоянно изменяется. Она может повышаться при стрессе или снижаться с уменьшением концентрации эстрогенов у женщин.
Лечение болезни Виллебранда разделяют на специфическое и неспецифическое. Наиболее важным патогенетически обусловленным моментом, является введение гемопрепаратов, содержащих комплекс фактора VIII и фактора Виллебранда. Открытым остается вопрос, какие кровотечения следует лечить с помощью заместительной терапии и какая должна быть доза препаратов с фактором Виллебранда? Трансфузию следует начинать за несколько дней до предполагаемых хирургических операций, так как коррекция гемостаза происходит постепенно. Переливание тромбоцитарной массы или применение препаратов, воздействующих на тромбоциты, не эффективно, так как в данном случае дисфункция тромбоцитов возникает вторично. При легкой и средней степени тяжести и развитии кровотечений микроциркуляторного типа доказана эффективность аминокапроновой кислоты.
В настоящее время в России действует программа «7 нозологий», которая предназначена для лечения больных семью редкими и дорогостоящими нозологиями за счет средств федерального бюджета. Благодаря этой программе значительно сократился процент инвалидизации, улучшилось качество жизни пациентов и снизился риск жизнеугрожающих кровотечений. Преимуществом программы «7 нозологий» стало предоставление лекарственной помощи по нозологическому принципу, а не по статусу инвалидности. Крайне важно сохранить действующую систему лекарственного обеспечения и централизованную закупку препаратов, так как для некоторой категории пациентов эта программа становится единственной возможностью поддерживать и сохранять собственную жизнь.
Список используемой литературы:
Christine A. Lee, Rezan A. Kadir, Peter A. Kouides: Inherited Bleeding Disorders in Women.
- Chandler WL, Peerschke EI, Castellone DD,
Meijer P (June 2011). «Von Willebrand factor assay proficiency testing.
The North American Specialized Coagulation Laboratory Association
experience». Am. J. Clin. Pathol. 135 (6): 862–9.
- «Molecular basis of von Willebrand
disease and its clinical implications». Haematologica 89 (9): 1036. 1
September 2004.
Подготовила: Драпкина Ю.С.
Болезнь фон Виллебранда : Генетические заболевания : Все про гены!
Фактор Виллебраннда — это мультимерный гликопротеин, который необходим для адгезии тромбоцитов (прилипание, прикрепление тромбоцитов к сосудистой стенки в зоне повреждения целостности эндотелия). Известно, что это заболевание встречается не только у людей, но и у собак (в частности, доберманов и пинчеров) и редко у свиней, крупного рогатого скота, лошадей и кошек. Существуют четыре типа наследственной болезни Виллебранда. На течение заболевания значительно влияют другие факторы, особенно группа крови.Признаки и симптомы
Риск возникновения кровотечения при заболевании БВ отличается в зависимости от типа болезни. Чаще основными проявлениями расстройства являются: беспричинное появление синяков, периодические носовые кровотечения и кровоточивость десен. У женщин может возникать меноррагия (усиление и увеличение продолжительности менструальных кровотечений), а во время родов больные женщины, могут страдать от избыточной потери крови. Иногда (в основном при заболевании 3 тип БВ), заболевания вызывает тяжелые внутренние кровотечения или скопление крови в суставах (гемартроз) вследствие относительно незначительных травм.
Диагностика
Если у лица есть подозрение на наличие болезни фон Вилебранда, то для точной диагностики необходимо провести количественное и качественное исследование плазмы крови пациента на наличие фактора фон Виллебранда. Этот процесс осуществляется путем измерения уровня фактора фон Виллебранда (антигенов этого фактора), содержащихся в исследуемом образце крови. Касательно проверки функциональности этого фактора, то обычно определяют уровень связывания фактора Виллебранда с гликопротеином (GP) Ib, его связь с коллагеном. Другими методами проверки активности ФВ может быть определение активности кофактор ристоцетина (RiCof) или скорость агглютинации тромбоцитов, после введения ристоцетина (ristocetin induced platelet agglutination (RIPA)).
Кроме того, может быть осуществлена проверка уровня фактора VIII, ведь фактор фон Виллебранда препятствует быстрому разрушению фактора VIII в крови. То есть, дефицит фактора фон Виллебранда может привести к резкому снижению уровня фактора VIII. Однако, даже нормальный уровень этих коагуляционных факторов, не исключает наличие заболевания, особенно если речь идет о 2 тип БВ. В таком случае, болезнь может быть обнаружена только путем исследования взаимодействия тромбоцитов с субэндотелиальным слоем в потоке крови (фактор активации тромбоцитов PAF), что проводится только в узкоспециализированных лабораториях (изучающие нарушения коагуляции) и обычно не осуществляется в большинстве обычных медицинских лабораторий. Анализ агрегации тромбоцитов, как правило, показывает ненормальную реакцию на введение ристоцетина, однако реакция на другие антагонисты будет нормальной. При проведении исследования с помощью аппарата PFA 100, (который позволяет выявить дефекты адгезии тромбоцитов к коллагене), время прекращения кровотечения при изучении реакции коллаген / адреналин будет нарушенным, но, время реакции коллаген / АДФ (как правило) находится в пределах нормы. Тип 2N болезни фон Виллебранда может быть диагностирован только после проведения анализа активности фактора VIII. Выявление БВ осложняется еще и тем, что фактор фон Виллебранда — это острофазовый белок, то есть его уровень в организме резко повышается во время беременности, при стрессе и инфекционных заболеваниях.
Одним из обязательных анализов, который должен проводиться при диагностике пациентов, у которых возникают внезапные кровотечения — это полный анализ крови (особое внимание при этом следует обратить на уровень тромбоцитов). Кроме того, иногда определяют: АЧТВ (активированное частичное тромбопластиновое время), протромбиновое время, тромбиновое время и уровень фибриногена. Если врачи подозревают заболевание гемофилией В, то осуществляется тестирование, которое определяет уровень активности фактора IX. В зависимости от результатов вышеперечисленных скрининговых тестов, может быть осуществлено исследование функциональности других факторов свертывания крови. У лиц с болезнью фон Виллебранда, как правило, нормальное протромбиновое время, однако тромбопластиновое время несколько больше нормы.
|
Заболевание |
Протромбированое время |
Активированное частичное тромбопластиновое время |
Время свертывания крови |
Уровень тромбоцитов |
|
Дефицит витамина К или прием варфарина |
продлен |
продлен |
нормальный |
нормальный |
|
ДВЗ синдром |
продлен |
продлен |
продлен |
уменьшен |
|
Хвороба фон Віллебранда |
нормальный |
продлен |
продлен |
нормальный |
|
Гемофілія |
нормальный |
продолен |
нормальный |
нормальный |
|
Побічні ефекти дії аспірину |
нормальный |
нормальный |
продлен |
нормальный |
|
Тромбоцитопенія |
нормальный |
нормальный |
продлен |
уменьшен |
|
Початкова стадія печінкової недостатності |
продлен |
нормальный |
нормальный |
нормальный |
|
Кінцева стадія печінкової недостатності |
продлен |
продлен |
продлен |
уменьшен |
|
Уремія |
нормальный |
нормальный |
продолен |
нормальный |
|
Вроджена афібріногенемія |
продлен |
продлен |
продлен |
нормальный |
|
Дефіцит фактору V |
продлен |
продлен |
нормальный |
нормальный |
|
Дефіцит фактору Х, наприклад при амілоїдозній пурпурі |
продлен |
продлен |
нормальный |
нормальный |
|
Тромбастенія Гланцмана |
нормальный |
нормальный |
продлен |
нормальный |
|
Синдром Бернара-Сульє (Bernard Soulie) |
нормальный |
нормальный |
продлен |
уменьшен |
Типы и классификация
Классификация
На сегодня известно четыре типа наследственной болезни Виллебранда — тип 1, тип 2, тип 3, и тромбоцитарный тип. Существуют врожденные и приобретенные формы БВ. В большинстве случаев болезнь фон Виллебранда — это наследственное расстройство, однако известны также случаи, возникновения заболевания в течение жизни. Международное общество по тромбоза и гемостаза (ISTH) классифицирует болезнь фон Виллебранда согласно определению качественного и количественного дефектов фактора фон Виллебранда.
Тип 1 БВ (60-80% всех случаев заболевания) — это количественное нарушение, то есть заболевание вызвано недостаточным уровнем фактора фон Виллебранда (этот тип гетерозиготный за дефектным геном), однако при заболевании на этот тип существенных нарушений процесса свертывания крови нет, именно поэтому большинство больных, обычно живут почти нормальной жизнью. Проблемы могут возникнуть только в виде кровотечения после операций (в том числе, во время стоматологических процедур), кроме того, могут появляться синяки (даже от легкого нажатия), для женщин характерно явление меноррагии. Наблюдается снижение уровня фактора Виллебранда на 10-45% от нормы (то есть его уровень составляет 10-45 IU МО). Тип 2 БВ (20-30% всех случаев заболевания) — это качественное нарушение, при нем риск возникновения внезапного кровотечения зависит от конкретного случая. Однако, уровень фактора Виллебранда при этом — нормальный, но структура мультимерного гликопротеина – нарушена или в больших или малых мультимерах отсутствуют определенные субъединицы. Выделяют четыре подтипа 2 типа заболевания фон Виллебранда: 2А, 2В, 2М и 2N. Тип 2A
Возникает при нарушении синтеза или протеолиза мультимера, составляющих фактор фон Виллебранда, в результате чего в крови присутствуют его малые мультимерние субъединицы. Связывание фактора VIII является нормальным. Зато, активность кофактор ристоцетина — очень низкая, по сравнению с антигеном фактора фон Виллебранда.
Тип 2B
При подтипы 2B отмечается повышенное родство фактора фон Виллебранда к рецептору на мембране тромбоцитов гликопротеина Ib, что приводит к быстрому выведению из организма тромбоцитов и больших мультимер ФВ.
Для этого типа характерно незначительная тромбоцитопения. Большие мультимеры фактора фон Виллебранда в кровообращении отсутствуют, однако активность фактора VIII находится в пределах нормы. Это можно объяснить тем, что крупные мультимеры Виллебранда образуют связь с тромбоцитами больного человека. Больные этим типом не могут использовать десмопрессин для остановки кровотечения, ведь этот препарат может усилить агрегацию тромбоцитов.
Тип 2М
Тип 2М болезни фон Виллебранда вызванный качественным дефицитом фактора фон Виллебранда. Несмотря на нормальный уровень антигена, функциональность фактора — снижается (снижение RICOF), что и отличает этот тип от типа 2А, ведь функциональный дефицит возникает не вследствие отсутствия или изменения размеров мультимера.
Тип 2N (Нормандия)
Это дефицит, который приводит к нарушению связывания фактора Виллебранда с фактором VIII. При заболевании на этот тип, у людей уровень антигена фактора фон Виллебранда — нормальный, равно как и не нарушена его функциональность. Однако, уровень фактора VIII значительно ниже нормы. Именно поэтому, вероятно, раньше пациентам, больным типом 2N часто ставили диагноз гемофилия А. Поэтому, если есть подозрения того, что у больного гемофилия А, особое внимание следует уделить семейной истории заболеваний и определить возможное наследование Х-сцепленной или аутосомной болезни .
Тип 3
Тип 3 — это самая тяжелая форма болезни фон Виллебранда (гомозиготная за дефектным геном). При заболевании на нее, у бльных могут возникать серьезные кровотечения из слизистых оболочек. У них, как правило, полностью отсутствует антиген фактора Виллебранда, а уровень фактора VIII очень низкий. Кроме того, у больных этим типом, часто возникает гемартроз (скопление крови в суставах), как и в случаях легкой формы гемофилии.
Тромбоцитарный тип Тромбоцитарный тип (также известный как псевдо-болезнь фон Виллебранда) — это аутосомно-доминантный тип БВ вызван усилением мутаций гена, кодирующего деятельность тромбоцитарного рецептора фактора Виллебранда, а именно, альфа-цепи рецептора гликопротеина Ib (GPIB). Этот белок является частью большего комплекса (GPIB / V / IX), который формирует полный рецептор фактора Виллебранда на тромбоциты. Активность ристоцетина и потеря больших мультимер Виллебранда, делает этот тип похожим на тип 2B, но генетическое тестирование фактора Виллебранда не обнаружит никаких мутаций.
Приобретенная форма болезни фон Виллебранда
Как уже было сказано выше, болезнь фон Виллебранда может быть не только наследственной переданной, но и возникнуть в течение жизни. Однако приобретенная форма болезни фон Виллебранда характерна только для тех лиц, в организме которых есть аутоантитела (антитела, способные взаимодействовать с аутоантигенами, т.е. с антигенами собственного организма, могут образовываться спонтанно или вследствие перенесенных инфекций). При этом типе заболевания, активность фактора фон Виллебранда не снижена, однако комплекс антител соединенных с ФВ очень быстро выводится из организма.
Обычно на этот тип расстройства болеют пациенты со стенозом аортального клапана, в результате действия заболевания возникают желудочно-кишечные кровотечения (синдром Гейда). Согласно мнениям исследователей — эта форма заболевания сегодня наиболее распространена. Приобретенная форма БВ может также проявляться при таких болезнях как опухоль Вильмса, гипотиреоз или мезенхимальных дисплазиях.
Патофизиология
Фактор фон Виллебранда, как известно, активируется за условие сильного кровотока и в условиях смещения тканей (shear stress). Именно поэтому, дефицит этого фактора свертывания сильнее влияет на органы с развитой сетью маленьких сосудов, такие как кожа, желудочно-кишечный тракт и матка. При ангиодисплазии (форма телеангиэктазии) толстой кишки, где напряжение сдвига значительно выше, чем в средних капиллярах, риск возникновения кровотечения соответственно, возрастает.
При тяжелых случаях типа 1 БВ, все генетические изменения происходят в гене, который кодирует функциональные характеристики фактора фон Виллебранда и характеризуются высокой панерантностю. При легких формах типа 1 БВ широкий спектр молекулярной патологии может возникать в дополнение к полиморфизмам гена. Группа крови человека (система АВО) также может влиять на течение заболевания, определяя ее симптомы и тяжесть воздействия на организм. Как правило, у лиц, с I (O) группой крови, средний уровень фактора фон Виллебранда ниже чем у людей с другими группами крови. Именно поэтому при исследовании уровня фактора фон Виллебранда с помощью системы ABO следует помнить, вышеуказанную особенность. Ведь известны такие случаи, когда здоровым людям с I (О) группой крови диагностировали I тип ХО, в то время, как лдей с IV (АВ) группой крови, у которых присутствовали определенные генетические нарушения считали вполне здоровыми, в связи с повышенным в этой группе крови естественным уровнем ФВ.
Генетика
Ген, кодирующий деятельность ФВ расположен на двенадцатой хромосоме (12p13.2). Он имеет 52 экзоны и охватывает 178kbp. 1 и 2 тип болезни фон Виллебранда наследуются по аутосомно-доминантному типу, в то время как 3 тип наследуется по аутосомно-рецессивному типу. Однако, известны случаи, когда 2 тип также наследовался рецессивно.
Эпидемиология
БВ встречается примерно у 1 человека со 100 . Однако у большинства из этих лиц никаких симптомов расстройства не проявляется. Частота клинически важных случаев — значительно ниже, в такой степени заболевание проявляется у 1 человека на 10000 жителей. Поскольку большинство форм БВ достаточно мягкие, тем чаще они оказываются у женщин, у которых увеличивается продолжительность и интенсивность менструаций. Обычно тяжелая форма заболевания, которая соответственно быстро проявляется, возникает у людей с I (О) группой крови.
Лечение
Пациентам с болезнью фон Виллебранда, как правило, ни какого регулярного лечения не требуется, хотя риск кровотечения у них всегда повышенный. Для женщин с тяжелыми менструальными кровотечениями, могут быть рекомендованы комбинированные оральные противозачаточные таблетки, которые, как правило, снижают кровотечения, сокращают продолжительность и частоту менструации. Иногда, больным БВ, которым планируется осуществлять хирургическую операцию проводять профилактическое лечение. В таком случае, людям обычно вводят препарат, основой которого является концентрат фактора VIII в комплексе с фактором фон Виллебранда (антигемофильный фактор, более известный как Humate-P). Если течение заболевания является умеренным, то профилактика может осуществляться с помощью десмопрессина (1-desamino- 8-D-аргинин вазопрессина, DDAVP) (десмопрессин ацетат, Stimate), который повышает уровень фактора Виллебранда в плазме пациента, путем высвобождения ФВ, который содержится в тельцах Вэйбел-Паладе, находящихся в эндотелиальных клетках.
История
Болезнь фон Виллебранда названа в честь Адольфа Эрика фон Виллебранда, финского педиатра, который впервые описал это заболевание в 1926 году.
Болезнь Виллебранда | Info-Farm.RU
Болезнь Виллебранда — наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирование, которое участвует в адгезии тромбоцитов на коллагене и защищает VIII фактор от протеолиза. При дефиците фактора Виллебранда, VIII Фактор подвергается протеолиза и его содержание в плазме снижается. Кроме того, при мин. Виллебранда снижается содержание серотонина и развивается патологическая дилятация сосудов и повышение их проницаемости. При мин. Виллебранда наблюдается длинные кровотечения т.к. у больных нарушены все три звена гемостаза.
Есть три формы болезни Виллебранда: унаследованный, приобретенный и псевдо или тромбоцитарный тип. Есть три типа наследственной болезни Виллебранда: ХВ Тип I, ХВ Тип II, и ХВ III. За три унаследованных типами ХВ существуют различные подтипы. Тромбоцитарный тип ХВ также наследственное заболевание. ХВ I типа является найпоширинишою и протекает, как правило, бессимптомно или проявляется легкими симптомами, такими как носовые кровотечения. Существуют различные факторы, которые влияют на презентацию и тяжесть симптомов ХВ, таких как тип крови.
Причина кровотечений — нарушение свертывания крови за недостаточной активности фактора Виллебранда. Распространенность болезни Виллебранда составляет 1 на 800-1000.
Болезнь Виллебранда названа в честь Эрика Адольфа фон Виллебранда, финского педиатра, впервые описал заболевание в 1926 году.
Синонимы
Ангиогемофилия; атромбопеничний пурпур; атромбоцитопеничний пурпур; геморрагическая капиляропатия; конституциональный тромбопатия, конституциональный тромбопатия фон Виллебранда-Юргенса; наследственная псевдогемофилия; синдром Юргенса; сосудистая гемофилия, псевдогемофилия.
Классификация
Различают три типа болезни Виллебранда.
- 1-й тип обусловлен частичным количественным дефицитом фактора Виллебранда. При этом его мультимерна структура сохранена. Имеется снижение прокоагулянтной активности фактора VIII, агрегации тромбоцитов, индуцированной ристоцетин, ристоцетинкофакторной активности, антигена фактора Виллебранда. Частота данной формы составляет от 75% до 80% всех случаев болезни Виллебранда. Наследование аутосомно-доминантное.
- 2-й тип обусловлен качественными изменениями фактора Виллебранда, связанный с нарушением формирования мультимера и подразделяется на подтипы: 2A, 2B, 2M, 2N.
- Фенотип подтипа 2A является результатом нарушения двух разных механизмов: дефекта синтеза высокомолекулярных мультимера и повышение протеолиза фактора Виллебранда. При подтипы 2B отмечается повышенное сродство фактора Виллебранда к рецептору на мембране тромбоцитов гликопротеина Ib.
- Подтип 2В .Это дефект «усиление функции». Способность качественно дефектного фактора фон Виллебранда привязываться к гликопротеину1 (GP1) рецептора на мембране тромбоцитов аномально повышенная, что приводит к его спонтанного связывания с тромбоцитами и дальнейшего быстрого клиренса из связанных тромбоцитов и больших мультимера фактора Виллебранда. Может возникнуть тромбоцитопения, а большие мультимера Виллебранда исчезнуть из циркуляции.
- Подтип 2M характеризуется нарушением связи фактора Виллебранда с рецептором гликопротеином Ib на мембране тромбоцитов.
- Подтип 2N характеризуется нормальным уровнем фактора Виллебранда и низкой прокоагулянтная активностью, что обусловлено нарушением связи фактора VIII и фактора Виллебранда.
Наследование болезни Виллебранда 2-го типа аутосомно-доминантное, за исключением подтипа 2N, где оно рецессивный. Частота появления данных форм составляет от 5% до 15% всех случаев болезни Виллебранда.
- 3-й тип — наиболее тяжелая форма с полным дефицитом фактора Виллебранда. Эта форма характеризуется отсутствием фактора Виллебранда в плазме, тромбоцитах и сосудистой стенке. Уровень фактора VIII ниже 10%. Наследование — аутосомно-рецессивное. Заболевание проявляется у гомозигот с одинаковыми дефектными аллелями или в двойных гетерозигот с двумя различными дефектными аллелями. У пациентов с 3-м типом имеется вероятность появления аллоантитил к фактору Виллебранда. Частота встречаемости заболевания 3-го типа болезни Виллебранда менее 5%.
Кроме того, существует тромбоцитарный тип болезни Виллебранда, который обусловлен мутацией в гене тромбоцитарного рецептора гликопротеина Ib, в результате которой повышается чувствительность данного рецептора к высокомолекулярных мультимера фактора Виллебранда. Фенотип аналогичный подтипа 2B.
- Приобретенный синдром Виллебранда определяется у пациентов с аутоиммунными, лимфопролиферативных заболеваниями, обусловлен появлением ингибитора против фактора Виллебранда, а также качественными аномалиями фактора VIII в связи с адсорбцией высокомолекулярных мультимера патологическими белками.
Приобретенная болезнь Виллебранда
Приобретенная болезнь Виллебранда может возникнуть у больных с аутоантителами. В этом случае функция фактора Виллебранда НЕ ингибированная, но комплекс фактор Виллебранда — антитело быстро выводится из кровотока.
Еще одна Форма ХВ возникает у пациентов с аортальным стенозом, что приводит к желудочно — кишечных кровотечений (синдром Хейди — англ. Heyde’s syndrome). В 2003 г.. Выяснилось, что пациенты с приобретенной болезнью Виллебранда и аортальным стенозом, перенесших протезирование клапанов, требовали коррекции аномалий гемостаза. Но эти аномалии могли проявляться повторно за 6 месяцев, если протез клапана плохо подходил пациенту. Кроме того, появляется склонность к кровотечениям у людей с имплантатом из левого желудочка (Left Ventricular Assist Device — LVAD), насос, качает кровь из левого желудочка сердца в аорту. В обоих случаях наблюдается разрушение крупных мультимера фактора Виллебранда, вследствие механических давления и нагрузок.
Тромбоцитемия является еще одной причиной приобретенной болезни Виллебранда, в связи с отторжением фактора Виллебранда через адгезию огромного числа тромбоцитов.
Приобретенная болезнь Виллебранда также была описана при следующих нарушениях: опухоли, гипотиреоз и мезенхимальные дисплазии Вильмса.
Патофизиология
ФВ в наиболее активен в условиях высокого кровотока и напряжения. Дефицит фактора Виллебранда, проявляется прежде всего в органах с большим количеством мелких сосудов, таких как кожа, желудочно — кишечный тракт и матка. В ангиодисплазии, форма телеангиэктазии толстой кишки, напряжение гораздо выше, чем в среднем в капиллярах и поэтому риск кровотечения увеличивается.
При тяжелых случаях типа 1 ХО, все генетические изменения происходят в гене, кодирующем функциональные характеристики фактора Виллебранда и характеризуются высокой пенетрантностью. При легких формах типа 1 ХВ широкий спектр молекулярной патологии может возникать в дополнение к полиморфизма гена. Группа крови человека (система АВО) также может влиять на течение заболевания, определяя ее симптомы и тяжесть воздействия на организм. Как правило, у лиц с I (O) группой крови, средний уровень фактора Виллебранда ниже чем у людей с другими группами крови. Именно поэтому при исследовании уровня фактора Виллебранда с помощью системы ABO следует помнить о вышеуказанную особенность. Ведь известны такие случаи, когда здоровым людям с I (О) группой крови диагностировали I тип ХВ, в то время, как лдей с IV (АВ) группой крови, в которых присутствовали определенные генетические нарушения считали вполне здоровыми, в связи с повышенным в этой группе крови естественным уровнем ФВ.
Генетика
Ген фактора Виллебранда находится на двенадцатой хромосоме (12p13.2). Он 52 экзоны, охватывающих 178kbp. Типы 1 и 2 наследуются как аутосомно-доминантный признак, а тип 3 имеет аутосомно-рецессивное наследование. Иногда тип 2 также наследуется рецессивно.
Эпидемиология
Распространенность ХВ составляет около 1 в 100 человек. Однако большинство из этих людей не имеют никаких симптомов. Распространенность клинически значимых случаев составляет 1 на 10 000. Поскольку большинство форм достаточно мягкие, они оказываются чаще у женщин, чья склонность к кровотечениям проявляется во время менструации. Эти проявления могут быть более тяжелыми и выраженными у людей с группой крови I (О).
Клиническая картина
Наиболее характерными и специфическими симптомом при болезни Виллебранда являются кровотечения из слизистых полости рта, носа, внутренних органов. Симптомы кровоточивости варьируют от умеренно выраженных до крайне тяжелых, протекающих преимущественно по микроциркуляторном типа. У пациентов с резким дефицитом фактора VIII наблюдаются обильные и длительные кровотечения (носовые, десневые, маточные), также кровоизлияния в мышцы и суставы. Кроме того, могут возникать длительные кровотечения при травмах, удалении зубов, операциях.
В детском возрасте часто бывают кровотечения из слизистых оболочек полости рта, носовые кровотечения, синяки на коже. Более тяжелое течение геморрагического диатеза отмечается во время или вскоре после перенесенных инфекционных заболеваний. Наиболее вероятным пусковым механизмом кровотечения на фоне инфекции является нарушение проницаемости сосудов. Вследствие этого появляются самопроизвольные кровотечения диапедезного типа.
Гематомы — кровоизлияния в подкожную клетчатку и мышечные ткани наблюдаются преимущественно после травм у больных с тяжелыми формами заболевания.
При болезни Виллебранда геморрагический синдром проявляется не всегда, периоды обострения чередуются с периодами повноги или почти полного отсутствия геморрагии. У некоторых пациентов болезнь Виллебранда может сочетаться с признаками мезенхимальные дисплазии: повышенной розтяжимистю кожи, слабостью связь с повышенной подвижностью суставов, пролапсом створок клапанов сердца.
Аутосомно тип наследования обусловливает одинаковую частоту возникновения болезни Виллебранда у пациентов обоих полов. У женщин в силу особенностей физиологического строения организма, связанных с репродуктивной функцией, наблюдается более частое проявление геморрагических симптомов. Около 65% женщин с болезнью Виллебранда страдают меноррагиями. Рецидивирующие маточные кровотечения, которые продолжаются более 10 дней, сопровождаются постгеморрагичною анемией.
Желудочно — кишечные кровотечения у пациентов с болезнью Виллебранда не является основной формой кровоточивости. Они могут быть вызваны приемом препаратов, влияющих на агрегацию тромбоцитов (ацетилсалициловая кислота и другие нестероидные противовоспалительные средства). Кроме того, источниками кровотечений являются латентные язвы желудка и двенадцатиперстной кишки, а также эрозивные гастриты, геморроидальные узлы.
У пациентов с болезнью Виллебранда могут быть длительные кровотечения при операциях, у женщин — во время родов. Роды у женщин с болезнью Виллебранда связаны с риском возникновения значительной кровопотери. У большинства пациентов со средней и легкими формами заболевания во время беременности уровень фактора VIII повышается в 2-3 раза и достигает нормальных значений, однако в послеродовом периоде возвращается к исходному уровню.
Гемартроз — наиболее редкий проявление болезни Виллебранда, характерное для заболевания 3-го типа. Острый гемартроз сопровождается болевым синдромом, обусловленным повышением внутрисуставного давления. Сустав увеличен в объеме, кожа над ним гиперемирована и горячая на ощупь. Если гемартроз возник после травмы, нужно исключить дополнительные повреждения (внутрисуставной перелом, отрыв отростка, ущемление тканей). Рецидивирующие гемартрозы вызывают хронический синовит. На стадии синовита синовиальная оболочка гипертрофируется и становится основным источником кровоизлияния в сустав. При остром синовите гемартрозы могут рецидивировать, несмотря на трансфузии фактора свертывания VIII, обусловлено воспалительным процессом в синовиальной оболочке. При хроническом синовите болевой синдром может отсутствовать, поскольку разрушена капсула сустава.
В отличие от гемофилии при болезни Виллебранда дальнейшего прогрессирования патологического процесса и развития деформирующего остеоартроза, как правило, не наблюдается.
Кровоизлияния в головной и спинной мозг и их оболочки при болезни Виллебранда возникают в связи с травмой. В отдельных случаях причиной таких кровоизлияний может быть гипертонический криз или прием препаратов, значительно нарушают гемостатическое функцию тромбоцитов (ацетилсалициловая кислота, бутадион и др.)
Учитывая аутосомно-доминантный тип наследования, генетический риск для потомства составляет 50% независимо от пола плода.
Диагностика
Диагностировать болезнь Виллебранда в легкой форме ее течения практически невозможно, поскольку кровотечения при ней возникают не часто. Человек может заметить только сильное кровотечение при тяжелой травме, или после хирургического вмешательства стоматолога.
Если у лица есть подозрение на наличие болезни Виллебранда, то для точной диагностики необходимо провести количественное и качественное исследование плазмы крови пациента на наличие фактора Виллебранда. Этот процесс осуществляется путем измерения уровня фактора Виллебранда (антигенов этого фактора), содержащихся в исследуемом образце крови. Касаясь проверки функциональности этого фактора, то обычно определяют уровень связывания фактора Виллебранда с гликопротеином (GP) Ib, его связь с коллагеном. Другими методами проверки активности ФВ может быть определение активности кофактора ристоцетин (RiCof) или скорость агглютинации тромбоцитов, после введения ристоцетин (ristocetin induced platelet agglutination (RIPA)).
Кроме того, может быть осуществлена проверка уровня фактора VIII, ведь фактор Виллебранда препятствует быстрому разрушению фактора VIII в крови. То есть, дефицит фактора Виллебранда может привести к резкому снижению уровня фактора VIII. Однако, даже нормальный уровень этих коагуляционных факторов не исключает наличие заболевания, особенно если речь идет о 2 тип ХВ. В таком случае болезнь может быть обнаружена только путем исследования взаимодействия тромбоцитов с субэндотелиальным слоем в кровотоке (фактор активации тромбоцитов PAF), что производится только в узкоспециализированных лабораториях и обычно не осуществляется в большинстве обычных медицинских лабораторий. Анализ агрегации тромбоцитов, как правило, показывает ненормальную реакцию на введение ристоцетин, однако реакция на другие антагонисты будет нормальной. Тип 2N болезни Виллебранда может быть диагностирован только после проведения анализа активности фактора VIII. Выявление ХВ осложняется еще и тем, что фактор Виллебранда — это острофазовый белок, то есть его уровень в организме резко повышается во время беременности, при стрессе и инфекционных заболеваниях.
Лечение
Пациентам с болезнью Виллебранда, как правило, никакого регулярного лечения не требуется, хотя риск кровотечения у них всегда повышенный. Для женщин с тяжелыми менструальными кровотечениями, могут быть рекомендованы комбинированные оральные противозачаточные, которые, как правило, снижают кровотечения, сокращают продолжительность и частоту менструации. Иногда, больным ХВ, которым планируется осуществлять хирургическую операцию проводят профилактическое лечение. В таком случае, людям обычно вводят препараты, основой которого является концентрат фактора VIII в комплексе с фактором Виллебранда (антигемофильный фактор, более известный как Humate — P). Если течение заболевания является умеренным, то профилактика может осуществляться с помощью десмопрессина (1 — desamino — 8 — D — аргинин вазопрессина, DDAVP) (десмопрессин ацетат, Stimate), который повышает уровень фактора Виллебранда в плазме пациента.
История
В 1924 году 5 — летняя девушка, которая жила на Аландских островах была доставлена в больницу в Хельсинки, Финляндия, где она была замечена доктором Эриком фон Виллебранда. Он, в конечном счете, обследовал 66 членов его семьи и сообщил в 1926 году, что он открыл ранее описанное нарушение свертываемости крови, отличающийся от гемофилии. Доктор фон Виллебранда определил аутосомно наследования и отметил, что симптомы кровотечения были более выражены у детей и женщин детородного возраста. Таким образом, он заявил, что пациенты с этим синдромом имели кожно — слизистые кровотечения, нормальное время свертывания, аутосомно наследования вместо связанного с Х — хромосомой и длительную кровотечение методом Дюка. Впоследствии он обнаружил, что переливание крови были действенными не только при коррекции анемии, но и для остановки кровотечений.
В 1950 году стало ясно, что у этих лиц был снижен «фактор плазмы», антигемофильный фактор (фактор VIII). С этого времени, фактор, вызывающий длительные кровотечения начал называться «фактор Виллебранда» в честь доктора Эрика фон Виллебранда.
В 1980-х, молекулярные и клеточные исследования отличили гемофилией А и болезнью Виллебранда точнее. Лица, которые болели ХВ имели нормальный ген фактора VIII на Х-хромосоме, но имели аномальный ген фактора Виллебранда на 12 хромосоме.
Болезнь Виллебранда — симптомы болезни, профилактика и лечение Болезни Виллебранда, причины заболевания и его диагностика на EUROLAB
. Впервые данное заболевание было изучено в 1926 г. ученым Виллебрандом, который на Аландских островах описал семью со своеобразным аутосомно-доминантно наследуемым геморрагическим диатезом, сходным как с тромбоцитовазопатией, так и с гемофилией. Было отмечено, что члены данной семьи страдают классической формой болезни, принадлежащей к I типу. Подтверждено доминантное наследование с разной проявляемостью патологического гена. Этим болезнь Виллебранда отличается от гемофилии, которая жестко наследуется и при которой у всех болеющих членов семьи в разных поколениях отмечается одна и та же выраженность дефицита фактора VIII.
По распространенности среди всех наследственных геморрагических диатезов болезнь Виллебранда занимает 3-е место после тромбоцитопатий и гемофилии А.
В основе патогенетических механизмов болезни Виллебранда лежит нарушение синтеза основного крупномолекулярного компонента фактора VIII, называемого также фактором Виллебранда.
По вышеперечисленным характеристикам различают «классический» тип болезни Виллебранда (тип I), при котором имеется более или менее выраженный парез синтеза рассматриваемого фактора с конкордантным снижением в плазме всех его компонентов и соответствующим отсутствием или уменьшением содержания в сосудистом эндотелии.
От этого типа отличаются вариантные формы болезни (тип II), при которых имеются качественные аномалии мультимерной структуры и свойств компонентов фактора VIII.
Болезнь по доминированию дефицита компонентов фактора VIII причисляется к плазменным дефектам гемостаза. При более углубленном рассмотрении с неменьшим правом ее можно отнести к первично-сосудистым заболеваниям, поскольку в основе болезни лежит ослабление или извращение синтеза фактора Виллебранда в эндотелии кровеносных сосудов — единственном месте его образования в организме.
Старое название болезни «ангиогемофилия» весьма близко к правильному пониманию ее сути, хотя в настоящее время редко употребляется.
Выраженность геморрагического синдрома при болезни Виллебранда варьирует от весьма легких форм с редко наблюдающимися носовыми кровотечениями и небольшими кровоизлияниями в кожу до крайне тяжелых вариантов с очень частыми, длительными и обильными кровотечениями самой разнообразной локализации, формированием гематом и больших кровоизлияний в мягких тканях и во внутренних органах. Иногда возникают кровоизлияния в суставы.
Геморрагический синдром при I типе намного тяжелее, чем при IIА и IIB типах болезни.
Следует отметить, что интенсивность кровотечений самой различной локализации (желудочно-кишечных, маточных, носовых) зачастую не соответствует нарушению коагуляциоиного и сосудисто-тромбоцитарного гемостазов.
В частности, на фоне умеренных нарушений в этих звеньях гемостаза упорно повторяются катастрофические кровотечения какой-либо одной локализации. В подобных случаях следует думать о каких-то дополнительных местных сосудистых или стромальных дисплазиях, провоцирующих кровотечения. Для их выявления необходимо тщательное дополнительное исследование слизистых оболочек носа, зева и глотки, ротовой полости, желудка и кишечника (риноскопия, ларингоскопия, фибродуоденогастроскопия, колоноскопия). На слизистых оболочках нередко обнаруживаются сосудистые образования в виде поверхностно расположенных расширенных и извитых сосудов диаметром 1-2 мм, легко дающих обильные, трудно останавливаемые кровотечения.
Известно, что такие сосудистые образования, чаще — артериовенозные соустья, служат причиной повторяющихся желудочно-кишечных кровотечений. Данные соустья наиболее опасны при болезни Виллебранда, когда нарушены основные механизмы купирования кровотечений.
Следует отметить, что сочетание болезни Виллебранда с ангиодисплазиями и другими дефектами соединительной ткани нельзя считать случайным. У больных с этим заболеванием часто выявляется пролабирование створок митрального и других клапанов сердца, ошибочно диагностируемое без эхокардиографии как ревматический митральный порок сердца.
В таком же свете следует рассматривать сочетания болезни Виллебранда с гиперэластозом кожи, слабостью связочного аппарата (частые и привычные вывихи, разболтанность суставов, реже — синдром Марфана).
Интенсивность кровотечения имеет прямую зависимость от уровня фактора VIII в плазме, что следует учитывать как при травмах, так и при хирургических вмешательствах.
При постановке диагноза заболевания Виллебранда основываются на совокупности следующих признаков: аутосомно-доминантном наследовании заболевания, кровоточивости, значительном удлинении времени кровотечения.
К основным диагностическим признакам добавляется ряд важных функциональных характеристик, облегчающих распознавание редуцированных вариантов болезни Виллебранда.
При болезни Виллебранда выявляется постепенное, а не немедленное нарастание активности фактора VIII в плазме больных после трансфузии антигемофильной плазмы.
Коррекционный эффект трансфузии намного превосходит количество вводимого фактора VIII.
Примечательна значительно большая, чем при гемофилии, продолжительность эффекта однократного переливания — около 36 ч, что характеризуется большей продолжительностью жизни в циркуляции реципиента фактора VIII.
Наиболее информативно количественное определение фактора Виллебранда в плазме больного.
Перечисленные диагностические признаки позволяют в большинстве случаев четко аргументировать диагноз болезни Виллебранда, определять ее тяжесть и форму.
Капилляроскопические изменения (неравномерность и закрученность петель капилляров, их булавовидные расширения) в диагностике болезни Виллебранда не используют, поскольку они выявляются менее чем у половины больных и далеко не патогномоничны, однако совместно с этим они весьма демонстративны и способствуют правильной диагностике.
Аутосомно-рецессивная форма болезни Виллебранда является самостоятельным заболеванием. У гетерозигот заболевание протекает совсем или почти бессимптомно, тогда как у гомозигот наблюдается крайне тяжелая кровоточивость при почти полном отсутствии фактора VIII в плазме. Однако поражение суставов и других частей опорно-двигательного аппарата, несмотря на такой значительный дефицит фактора VIII, все же гораздо легче, чем при гемофилии.
Наиболее важным патогенетическим моментом в терапии, способствующим нормализации (зачастую только временной) всех нарушенных гемостатических функций, является трансфузионная терапия – введение гемопрепаратов, содержащих комплекс фактора VIII, в том числе фактор Виллебранда.
С этой целью чаще всего используют антигемоф ильную плазму и криопреципитат.
Кроме того, под влиянием введенного извне фактора Виллебранда повышается собственная продукция фактора VIII, поэтому заместительная терапия болезни Виллебранда требует значительно более редких трансфузий и меньших доз гемопрепаратов, чем лечение гемофилии А. Однократные переливания антигемофильной плазмы или криопреципитата повышают к концу первых суток уровень фактора VIII почти до 100%, после чего его концентрация выше 50% поддерживается самостоятельно в течение 36 ч. Правда, концентрация самого фактора Виллебранда снижается раньше, в силу чего тромбоцитарно-сосудистый гемостаз вновь нарушается при еще высоком уровне VIIIk в плазме. Этим объясняется то, что при болезни Виллебранда трансфузионная терапия более стойко и надежно поддерживает уровень фактора VIII и предупреждает послеоперационные кровотечения, чем купирует микроциркуляторные кровотечения (маточные и носовые). В связи с этим наиболее целесообразно введение гемопрепаратов (антигемофильной плазмы, криопреципитата) не реже 1 раза в 2 дня и в разовой дозе не меньше 15 ЕД/кг.
Коррекция развивается постепенно, поэтому перед хирургическими вмешательствами трансфузии начинают за 2-4 дня до операции, а при родах – в самом начале родовой деятельности.
Показанием к заместительной терапии служат обильные и длительные кровотечения любой локализации, хотя при маточных кровотечениях такое лечение не всегда эффективно.
Переливания тромбоцитной массы при болезни Виллебранда неэффективны, поскольку дисфункция тромбоцитов при этой болезни вторична. По этой же причине при болезни Виллебранда оказались малоэффективными и многие фармакологические препараты, применяющиеся при тромбоцитопатиях, — АТФ, соли магния, серотонин и др.
Принципиально новым является использование в лечении болезни Виллебранда аргинин-терминального синтетического аналога вазопрессина.
При легких и среднетяжелых заболеваниях доказана эффективность аминокапроновой кислоты в дозах до 0,2 г/(кг/сут), которая применяется при всех кровотечениях микроциркуляторного типа, в том числе при маточных кровотечениях с первого дня менструального цикла до окончания менструации.
Следует избегать совместного применения аминокапроновой кислоты, противозачаточных препаратов и криопреципитата, так как такое лечение может осложниться диссеминированным внутрисосудистым свертыванием крови или тромбозами. Носовые кровотечения купируют так же, как при гемофилии.
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни Виллебранда, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
причины и развитие, типы, течение, диагностика, лечение
© Автор: Солдатенков Илья Витальевич, врач-терапевт, специально для СосудИнфо.ру (об авторах)
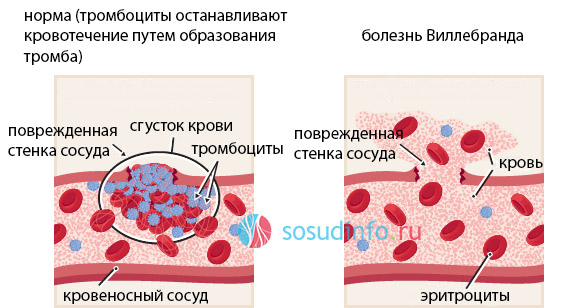
Болезнь Виллебранда (БВ, болезнь фон Виллебранда) — гематологическая патология, передающаяся по наследству и проявляющаяся внезапно возникающими кровотечениями. Дефицит фактора Виллебранда в крови нарушает работу всей свертывающей системы. VIII фактор подвергается протеолизу, кровеносные сосуды расширяются, их проницаемость повышается. Патология проявляется частыми кровотечениями различной локализации и интенсивности.
Гемостаз обеспечивается адекватной работой свертывающей системы крови и является защитной реакцией организма. При повреждении кровеносного сосуда начинается кровотечение. Активизируется система гемостаза. Благодаря плазменным факторам крови происходит агрегация и адгезия тромбоцитов, формируется сгусток, который закрывает имеющийся дефект в эндотелии. Дефицит хотя бы одного из факторов крови нарушает адекватный гемостаз.
Фактор Виллебранда (ФБ) — специфический белок системы гемостаза, отсутствие или недостаток которого приводит к нарушению процессов коагуляции. Этот мультимерный гликопротеин является активным носителем элемента VIII фактора, обеспечивает адгезию тромбоцитов, их прикрепление к сосудистой стенки в зоне повреждения эндотелия. Гликопротеин синтезируется в эндотелиоцитах и соединяет рецепторы тромбоцитов с субэндотелием. Болезнь передается от родителей к детям каждое поколение и чаще встречается у женщин.
Данная патология впервые была описана в начале прошлого столетия финским ученым Виллебрандом. Он наблюдал за семьей, члены которой страдали геморрагическим диатезом, сходным с гемофилией. Кровотечения у них протекали по гематомному типу, имели сложную форму и ставили под угрозу жизнь больных. Было доказано доминантное наследование патологии с разной проявляемостью патологического гена.
Заболевание имеет несколько наименований, но самым информативным является термин «ангиогемофилия». Он позволяет понять суть патологического процесса, но в настоящее время редко употребляется.
Раньше пациенты с болезнью Виллебранда рано становились инвалидами и редко доживали до зрелого возраста. В настоящее время больные с геморрагическим диатезом могут вести полноценный образ жизни, заниматься трудовой деятельностью и даже некоторыми малотравматичными видами спорта.
Классификация
Болезнь Виллебранда подразделяется на три типа:
- 1 тип — недостаточное содержание ФВ в крови, приводящее к снижению активности VIII фактора и нарушению агрегации тромбоцитов. Эта «классическая» форма патологии встречается чаще остальных. Частично или полностью блокируется синтез рассматриваемого фактора в сосудистом эндотелии. При этом работа свертывающей системы крови существенно не изменяется. Больные чувствуют себя удовлетворительно. Проблемы в виде кровотечений возникают после операций и стоматологических процедур. У них быстро появляются синяки даже от обычного прикосновения.
- 2 тип — ФВ находится в крови в нормальном количестве, изменяется его структура. Под воздействием провоцирующего фактора возникают внезапные кровотечения различной локализации и степени интенсивности.
- 3 тип — самая тяжелая форма патологии, обусловленная полным отсутствием ФВ в крови. Это очень редкая форма болезни, проявляющаяся микроциркуляторными кровотечениями и скоплением крови в суставных полостях.
- В отдельную группу выделяют тромбоцитарный тип, в основе которого лежит мутация гена, ответственного за состояние тромбоцитарного рецептора фактора Виллебранда. Тромбоцитарный фактор Виллебранда высвобождается из активных тромбоцитов и обеспечивает их адгезию и агрегацию.
Существует приобретенная форма патологии, которая встречается крайне редко. Механизм ее образования обусловлен появлением в крови аутоантител. Клетки собственного организма начинают восприниматься как чужеродные, к ним и вырабатываются антитела. Спровоцировать развитие патологии у лиц из группы риска могут острые инфекционные заболевания, травмы, стрессы. Этот тип БВ обнаруживают у пациентов с аутоиммунными заболеваниями, онкообразованиями, пониженной функцией щитовидной железы, мезенхимальными дисплазиями.
Причины
Болезнь фон Виллебранда – геморрагический диатез, при котором процесс свертывания крови полностью или частично нарушается. Гемостаз является довольно сложным процессом и состоит из нескольких стадий, последовательно сменяющих друг друга. Под воздействием определенных факторов свертывания начинается процесс тромбообразования, в результате которого образуется кровяной сгусток, закупоривающий место повреждения сосуда. При БВ снижается в крови содержание особого белка – фактора Виллебранда, который обеспечивает агрегацию тромбоцитов и их прилипание к поврежденному эндотелию.
 Главная причина болезни – полиморфизм гена, кодирующего синтез фактора Виллебранда. В результате он синтезируется в недостаточном количестве или вообще отсутствует в крови. БВ встречается как среди мужчин, так и среди женщин. В связи с физиологическими особенностями строения женского организма, обусловленными репродуктивной функцией, геморрагический синдром проявляется у женщин наиболее часто.
Главная причина болезни – полиморфизм гена, кодирующего синтез фактора Виллебранда. В результате он синтезируется в недостаточном количестве или вообще отсутствует в крови. БВ встречается как среди мужчин, так и среди женщин. В связи с физиологическими особенностями строения женского организма, обусловленными репродуктивной функцией, геморрагический синдром проявляется у женщин наиболее часто.
БВ часто протекает легко и может быть вообще не диагностирована. Дефицит ФВ обычно заканчивается кровотечениями из органов, имеющих развитую капиллярную сеть – кожу, ЖКТ и матку. Тяжелая форма заболевания, проявляющаяся клинически, возникает у людей с I (О) группой крови. Банальное кровотечение из носа или из лунки после удаления зуба может закончиться смертью больного.
Симптоматика
У здоровых людей при повреждении кровеносного сосуда мелкие кровяные пластинки направляются в очаг кровотечения, склеиваются друг с другом и закрывают образовавшийся дефект. У больных этот процесс нарушается, и кровь теряет способность сворачиваться.
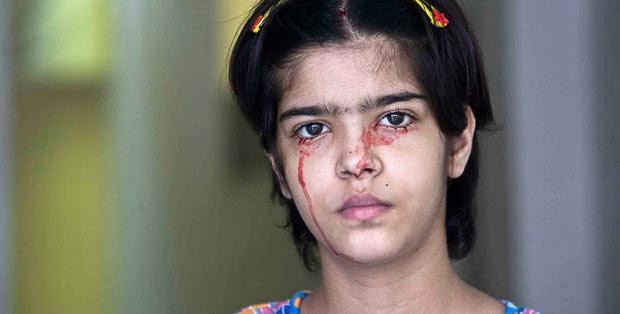
Специфическим симптомом заболевания является кровотечение различной интенсивности, обширности и локализации. Причины длительных кровотечений — травматические повреждения, оперативные вмешательства, стоматологические манипуляции. При этом у больных появляется слабость, головокружение, бледность кожи, учащается сердцебиение, снижается кровяное давление, возникает предобморочное состояние. Клиническая картина патологии во многом определяется величиной и скоростью кровопотери.
У детей геморрагический диатез протекает наиболее тяжело после перенесенных ОРЗ и прочих острых инфекций. Во время интоксикации повышается проницаемость сосудов, что приводит к появлению спонтанных кровотечений. Болезнь Виллебранда является неизлечимой патологией с волнообразным течением, при котором периоды обострения сменяются полным отсутствием геморрагий.
Основные проявления геморрагического синдрома при болезни Виллибранда:
- Кровотечения из пищеварительного тракта возникает после приема лекарств из группы НПВС и дезагрегантов. У больных обычно кровоточат язвы слизистой ЖКТ и геморроидальные узлы. Артериовенозные соустья часто становятся причиной повторяющихся кровотечений. Симптомами желудочного кровотечения является мелена – дегтеобразный черный жидкий стул и рвота измененной темной кровью.
- Гемартроз — кровоизлияние в полость сустава, проявляющееся болью, ограничением функции, отечностью и покраснением кожи, усилением болезненности при пальпации. Сустав увеличивается в объеме, становится шарообразным, контуры его сглаживаются. При продолжающимся кровотечении в сустав кожа становится синюшной, мягкие ткани – тугими, напряженными, появляется местная гипертермия.
- В особо тяжелых случаях геморрагический синдром у больных сочетается с признаками мезенхимальной дисплазии. Местные сосудистые и стромальные дисплазии провоцируют упорно повторяющиеся кровотечения преимущественно одной локализации.
Течение болезни Виллебранда непостоянно и изменяется со временем. Симптомы могут надолго исчезать и появляться вновь безо всяких на то причин. Некоторые пациенты спокойно живут с данной патологией и чувствуют себя удовлетворительно. Другие страдают от постоянных, смертельно опасных кровотечений. У них снижает качество жизни с самого рождения. Кровотечения возникают внезапно, являются массивными, останавливаются только в условиях стационара.
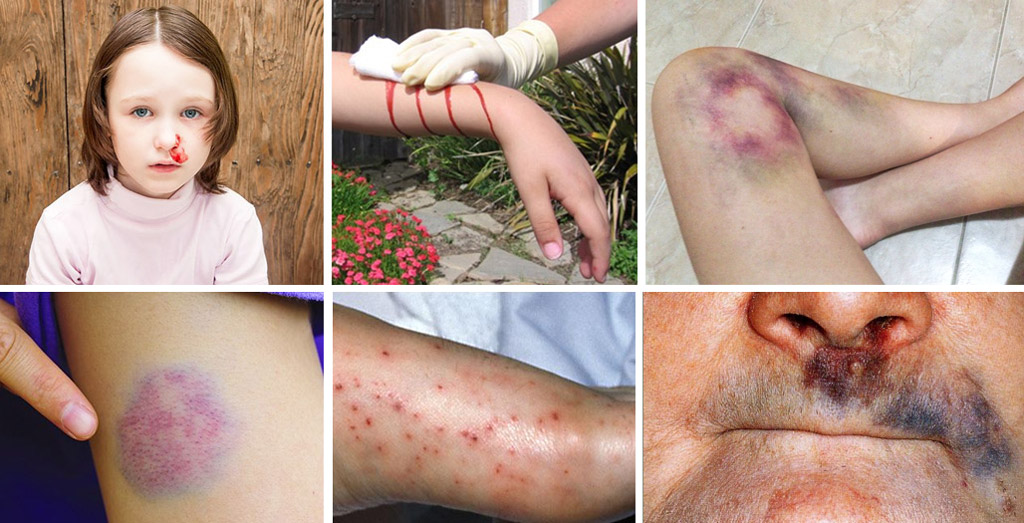
Симптоматика легкой формы патологии:
- Частые кровотечения из носа,
- Обильные менструальные кровотечения,
- Продолжительные кровотечения при незначительных повреждениях кожи,
- Петехии,
- Геморрагии после травм.
Клинические признаки тяжелой формы:
- Кровь в моче сопровождается болью в пояснице и симптомами дизурии,
- Обширные гематомы после легкого ушиба сдавливают крупные сосуды и нервные стволы, что проявляется болью,
- Гемартрозы, сопровождающиеся болью в пораженном суставе, его отеком, местной гипертермией,
- Продолжительные кровотечения из десен после чистки зубов,
- Кровотечения из зева и носоглотки могут привести к бронхообструкции,
- Кровоизлияния в оболочки мозга приводят к поражению ЦНС или летальному исходу.
В этом случае симптомы болезни практически идентичны гемофилии.
Диагностика
Болезнь Виллебранда трудно диагностировать. Чаще всего ее обнаруживают только в подростковом возрасте. Диагностика болезни Виллебранда начинается со сбора семейного анамнеза и опроса больного. Наследственная предрасположенность и выраженный геморрагический синдром — признаки, позволяющие врачу поставить предварительный диагноз.
Диагностические мероприятия при БВ:
- Медико-генетическое консультирование показано всем супружеским парам, находящимся в группе риска. Генетики выявляют носительство дефектного гена, анализируют генеалогические данные.
- Лабораторное определение активности фактора Виллебранда, его количества в плазме крови и функциональности.
- Анализ коагулограммы.
- Полный анализ крови — обязательный анализ в диагностике патологии. В общем анализе крови выявляют признаки постгеморрагической анемии.
- Выявить гемартрозы можно путем проведения рентгенографии суставов, диагностической артроскопии, компьютерной или магнитно-резонансной томографии; внутренние кровотечения — с помощью УЗИ брюшной полости, лапароскопии, эндоскопии; наружные кровотечения видны невооруженным глазом.
- Анализ кала на скрытую кровь.
- Проба жгута и щипка.
Лечение
Лечением болезни Виллебранда занимаются врачи-гематологи. Справиться с патологий окончательно невозможно, поскольку она имеет наследственный характер. Врачи борются с ее последствиями и облегчают жизнь больным.
Основу терапии составляет заместительная трансфузионная терапия. Она направлена на нормализацию всех звеньев гемостаза. Больным вводят гемопрепараты, содержащие фактор Виллебранда — антигемофильную плазму и криопреципитат. Заместительная терапия способствует повышению биосинтеза дефицитного фактора в организме.
 Остановить небольшое кровотечение поможет давящая повязка, гемостатическая губка, обработка раны тромбином.
Остановить небольшое кровотечение поможет давящая повязка, гемостатическая губка, обработка раны тромбином.- Кровоостанавливающим эффектом обладают лекарственные препараты: «Десмопрессин», антифибринолитики, гормональные пероральные контрацептивы при маточным кровотечениях.
- На кровоточащую рану наносят фибриновый гель.
- При гемартрозе накладывают на ногу гипсовую лонгету, прикладывают холод и придают конечности возвышенное положение. В дальнейшем больным назначают УВЧ и ограничение нагрузки на сустав. В тяжелых случаях проводят пункцию сустава под местной анестезией.
 Остановить небольшое кровотечение поможет давящая повязка, гемостатическая губка, обработка раны тромбином.
Остановить небольшое кровотечение поможет давящая повязка, гемостатическая губка, обработка раны тромбином.Для лечения болезни крови 1 и 2 типа применяют «Десмопрессин» – препарат, стимулирующий выброс в системный кровоток ФВ. Его выпускают в форме назального спрея и раствора для инъекций. Когда этот препарат оказывается неэффективным, проводят заместительную терапию плазменным концентратом ФВ.
К антифибринолитикам относятся аминокапроновая и транексамовая кислоты. Их вводят внутривенно капельно или принимают внутрь. Препараты на основе этих кислот наиболее эффективны при маточных, желудочно-кишечных и носовых кровотечениях. «Транексам» – основное средство в лечении легкой формы БВ. В тяжелых случаях препарат применяют в сочетании со специфическими гемостатиками – «Этамзилатом» или «Дициноном».
Профилактика
Предотвратить развитие болезни невозможно, поскольку она передаются по наследству. Снизить риск кровотечений можно путем соблюдения следующие профилактических мероприятий:
- Генетическое консультирование супружеских пар, входящих в группу риска,
- Диспансерное наблюдение за больными детьми,
- Регулярное посещение специализированного гематологического центра,
- Предупреждение травматизма,
- Запрет на прием «Аспирина» и прочих препаратов, снижающих функцию тромбоцитов,
- Проведение операций строго по жизненным показаниям,
- Ведение здорового образа жизни,
- Правильное питание.
Все эти мероприятия позволяют избежать появления внутрисуставных и внутримышечных кровотечений и предотвратить развития осложнений. Своевременная и адекватная терапия делает прогноз заболевания благоприятным. Тяжелое течение БВ с частыми и массивными кровотечениями ухудшает прогноз и состояние больных.
Видео: Лекция по болезни Виллебранда
Видео: болезнь Виллебранда в программе “Жить здорово”
Вывести все публикации с меткой:Рекомендации читателям СосудИнфо дают профессиональные медики с высшим образованием и опытом профильной работы.
На ваш вопрос ответит один из ведущих авторов сайта.
В данный момент на вопросы отвечает: А. Олеся Валерьевна, к.м.н., преподаватель медицинского вуза
Поблагодарить специалиста за помощь или поддержать проект СосудИнфо можно произвольным платежом по ссылке.