Митохондриальные заболевания — Википедия
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК[1].
Можно выделить две группы митохондриальных заболеваний:
- Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
- Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Наследование митохондриальных болезней[править | править код]
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все митохондрии наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между её потомками. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
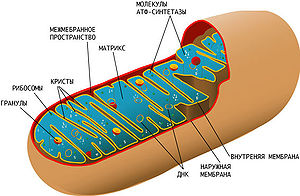 Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеваний
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеванийДефекты и симптомы[править | править код]
В общем случае митохондриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани,[2] поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Помимо относительно распространённой митохондриальной миопатии, встречаются:
- митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
- наследственная оптическая нейропатия Лебера (en:Leber’s hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде;
- синдром Вольфа-Паркинсона-Уайта (en:Wolff-Parkinson-White syndrome) Синдром WPW не относится к митохондриальным миопатиям.
- рассеянный склероз и подобные ему заболевания;[источник не указан 3021 день]
- синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в конце первого года жизни, иногда — во взрослом возрасте. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
- нейропатия, атаксия, retinitis pigmentos и птоз en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, туннельное зрение и потеря зрения, птоз, деменция;
- митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200.[3] «Горячей точкой» в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов[4]. Также в качестве одного из методов применяются пируваты
В настоящее время проводятся экспериментальные работы по изучению возможности экстракорпорального (in vitro) оплодотворения с использованием химерной яйцеклетки, ядро которой получено из яйцеклетки пациентки с митохондриальным заболеванием, а цитоплазму из другой яйцеклетки от женщины с нормально функционирующими митохондриями (замена ядра)[6].
- ↑ Scarpulla R.C. Nuclear control of respiratory gene expression in mammalian cells (англ.) // J Cell Biochem. : journal. — 2006. — Vol. 97, no. 4. — P. 673—683. — PMID 16329141.
- ↑ Finsterer J. Hematological manifestations of primary mitochondrial disorders (англ.) // Acta Haematol. : journal. — 2007. — Vol. 118, no. 2. — P. 88—98. — doi:10.1159/000105676. — PMID 17637511.
- ↑ Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2008. — August (vol. 83, no. 2). — P. 254—260. — doi:10.1016/j.ajhg.2008.07.004. — PMID 18674747.
- ↑ «When Cells Stop Working» dated November 5, 2006 at Time Magazine
- ↑ Tanaka M., Nishigaki Y., Fuku N., Ibi T., Sahashi K., Koga Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases (англ.) : journal. — 2007. — doi:10.1016/j.mito.2007.07.002. — PMID 17881297.
- ↑ Hello mothers, hello father (англ.). The Economist (27 October 2012). Дата обращения 17 декабря 2013.
На русском языке[править | править код]
На английском языке[править | править код]
Митохондриальные заболевания — это… Что такое Митохондриальные заболевания?
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариотов, в частности — человека.
Электронномикроскопическая фотография, показывающая митохондрии человека в поперечном сеченииОбщие сведения
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК[1].
Можно выделить 2 группы митохондриальных заболеваний:
- Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
- «Вторичные митохондриальные заболевания», включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печеночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола ). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все они наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между ее потомками, а затем происходит удвоение ДНК. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
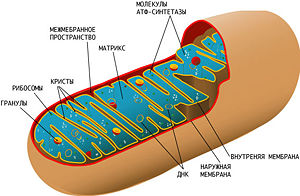 Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеваний
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеванийДефекты и симптомы
Эффекты митоходриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах, мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае, митоходриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани,[2] поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
- Митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
- наследственная оптическая нейропатия Лебера (en:Leber’s hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде;
- синдром Вольфа-Паркинсона-Уайта (en:Wolff-Parkinson-White syndrome)
- рассеянный склероз и подобные ему заболевания;[источник не указан 371 день]
- синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : После начала нормального постнатального развития организма болезнь обычно развивается в конце первого года жизни, но иногда проявляется у взрослых. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
- «Нейропатия, атаксия, retinitis pigmentos и птоз» en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, тунельное зрение и потеря зрения, птоз, деменция;
- Митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Диагностика
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
Эпидемиология
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200.[3] «Горячей точкой» в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов [4]. Также в качестве одного из методов применяются пируваты [5].
Поскольку в сперматозоиде, который вносит половину хромосом будущего организма, содержится мало митохондрий, митохондриальная наследственность определяется, в основном, митохондриями яйцеклетки. Сейчас проводятся экспериментальные работы по экстракорпоральному (in vitro) оплодотворению с использованием переноса ядра оплодотворённой яйцеклетки в безъядерную цитоплазму другой яйцеклетки с нормально функционирующими митохондриями (замена ядра).
См. также
Примечания
- ↑ Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006.97(4):673-83. PMID: 16329141
- ↑ Finsterer J (2007). «Hematological manifestations of primary mitochondrial disorders». Acta Haematol. 118 (2): 88–98. DOI:10.1159/000105676. PMID 17637511.
- ↑ Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF (August 2008). «Pathogenic mitochondrial DNA mutations are common in the general population». Am. J. Hum. Genet. 83 (2): 254–60. DOI:10.1016/j.ajhg.2008.07.004. PMID 18674747.
- ↑ «When Cells Stop Working» dated November 5, 2006 at Time Magazine
- ↑ Tanaka M, Nishigaki Y, Fuku N, Ibi T, Sahashi K, Koga Y (2007). «Therapeutic potential of pyruvate therapy for mitochondrial diseases». DOI:10.1016/j.mito.2007.07.002. PMID 17881297.
Ссылки
На русском языке
На английском языке
Классификация митохондриальных болезней | Компетентно о здоровье на iLive
Единой классификации митохондриальных заболеваний не существует из-за неопределённости вклада мутаций ядерного генома в их этиологию и патогенез. Существующие классификации основаны на 2 принципах: участии мутантного белка в реакциях окислительного фосфорилирования и кодируется ли мутантный белок митохондриальной или ядерной ДНК.
На основании двойственности кодирования митохондриальных белков процессов тканевого дыхания и окислительного фосфорилирования (ядерного и сугубо митохондриального) по этиологическому принципу выделяют 3 группы наследственных болезней.
- Митохондриальные болезни, обусловленные генными мутациями ядерной ДНК:
- дефекты транспортных субстратов;
- дефекты субстратов утилизации;
- дефекты ферментов цикла Кребса;
- нарушение окислительного фосфорилирования;
- нарушения в дыхательной цепи;. о дефекты импортации белков.
- Митохондриальные болезни, в основе которых лежат мутации митохондриальной ДНК:
- спорадические мутации;
- точковые мутации структурных генов;
- точковые мутации синтетических генов.
- Митохондриальные болезни, связанные с нарушением межгеномных сигнальных эффектов:
- множественные делеции митохондриальной ДНК, но наследуемые по аутосомно-доминантному типу;
- делеции (уменьшение количества) митохондриальной ДНК, наследуемые по аутосомно-рецессивному типу.
Выделяют также приобретённые митохондриальные болезни, связанные с воздействием токсинов, лекарств, старения.
К настоящему времени достаточно хорошо изучен патогенез митохондриальных болезней. В виде схемы его можно представить поэтапно следующим образом: транспорт субстратов, их окисление, цикл Кребса, функционирование дыхательной цепи, сопряжение тканевого дыхания и окислительного фосфорилирования. Транспорт субстратов осуществляется с помощью специальных транспортных белков — транслоказ, которые переносят дикарбоновые кислоты, АТФ, АДФ, ионы кальция, глутамат и др. Основные субстраты митохондрий — пируват и жирные кислоты, транспорт которых обеспечивает карнитин-пальмитоил-трансфераза и карнитин.
Окисление субстратов происходит при участии ферментов пируватдегидрогеназного комплекса, состоящего из 3 ферментов: пируватдегидрогеназы, липоат-ацетилтрансферазы и липоамид-дегидрогеназы с образованием ацетил-КоА, который включается в цикл Кребса. Утилизация жирных кислот происходит поэтапно в процессе бета-окисления. В ходе этих реакций образующиеся электроны переносятся в дыхательную цепь митохондрий. Полное разложение пирувата осуществляется в цикле Кребса, в результате которого образуются молекулы NAD и FAD, передающие свои электроны в дыхательную цепь. Последнюю образуют 5 мультиферментных комплексов, 4 из которых осуществляют транспорт электронов, а пятый катализирует синтез АТФ. Комплекс дыхательной цепи находится под двойным контролем ядерного и митохондриального геномов.
С позиций патогенеза можно выделить 3 основные группы митохондриальных заболеваний.
- Болезни процессов окислительного фосфорилирования.
- Болезни бета-окисления жирных кислот.
- Дефекты метаболизма пирувата и цикла Кребса.
С точки зрения ведущего биохимического дефекта митохондриальные болезни подразделяются на следующие группы.
- Дефекты транспорта субстратов.
- Дефицит монокарбокситранслоказы.
- Нарушение транспорта карнитин-ацилкарнитина (первичная мышечная недостаточность карнитина, системная недостаточность карнитина, смешанные формы дефицита карнитина, вторичная карнитиновая недостаточность, недостаточность карнитпальмитоилтрансферазы 1 и 2, комбинированная недостаточность карнитина и карнитин-пальмитоилтрансферазы).
- Дефекты утилизации субстратов.
- Дефекты окисления пирувата:
- недостаточность пируватдекарбоксилазы;
- недостаточность дигидролипоилтрансацетилазы;
- недостаточность дигидролипоилдегидрогеназы;
- недостаточность пируватдегидрогеназы;
- недостаточность пируваткарбоксилазы;
- недостаточность карнитин-ацетилтрансферазы.
- Дефекты окисления пирувата:
- Дефекты метаболизма свободных жирных кислот: дефекты бета-окисления жирных кислот.
- Дефекты дыхательной цепи.
- Дефекты NADH: KoQ-редуктазного комплекса (с нормальным уровнем карнитина и с карнитиновой недостаточностью).
- Дефекты KoQ цитохром Ь, cl-редуктазного комплекса (недостаточность KoQ-10, недостаточность Fe-S-протеинов, недостаточность цитохрома Ь, комбинированная недостаточность цитохромов b и cl).
- Недостаточность цитохрома а, аЗ.
- Недостаточность цитохрома а, аЗ и b.
- Дефекты накопления и передачи энергии.
- Нарушения окислительного фосфорилирования с гиперметаболизмом (болезнь Люфта).
- Нарушения окислительного фосфорилирования без гиперметаболизма.
- Недостаточность митохондриальной АТФазы.
- Недостаточность адениннуклеотидтранслоказы.
В настоящее время принята классификация по этиологическому принципу с выделением в каждой группе нескольких подгрупп заболеваний. Она наиболее обоснованна.
 [1], [2], [3], [4], [5], [6], [7], [8]
[1], [2], [3], [4], [5], [6], [7], [8]
Митохондриальные заболевания — Википедия
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека.
Общие сведения
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК[1].
Можно выделить две группы митохондриальных заболеваний:
- Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
- Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все митохондрии наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между её потомками. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеванийДефекты и симптомы
Эффекты митохондриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае митохондриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани,[2] поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
- митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
- наследственная оптическая нейропатия Лебера (en:Leber’s hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде;
- синдром Вольфа-Паркинсона-Уайта (en:Wolff-Parkinson-White syndrome) Синдром WPW не относится к митохондриальным миопатиям.
- рассеянный склероз и подобные ему заболевания;[источник не указан 2925 дней]
- синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в конце первого года жизни, иногда — во взрослом возрасте. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
- нейропатия, атаксия, retinitis pigmentos и птоз en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, туннельное зрение и потеря зрения, птоз, деменция;
- митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Диагностика
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
Эпидемиология
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200.[3] «Горячей точкой» в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов[4]. Также в качестве одного из методов применяются пируваты[5].
В настоящее время проводятся экспериментальные работы по изучению возможности экстракорпорального (in vitro) оплодотворения с использованием химерной яйцеклетки, ядро которой получено из яйцеклетки пациентки с митохондриальным заболеванием, а цитоплазму из другой яйцеклетки от женщины с нормально функционирующими митохондриями (замена ядра)[6].
См. также
Примечания
- ↑ Scarpulla R.C. Nuclear control of respiratory gene expression in mammalian cells (англ.) // J Cell Biochem. : journal. — 2006. — Vol. 97, no. 4. — P. 673—683. — PMID 16329141.
- ↑ Finsterer J. Hematological manifestations of primary mitochondrial disorders (англ.) // Acta Haematol. : journal. — 2007. — Vol. 118, no. 2. — P. 88—98. — DOI:10.1159/000105676. — PMID 17637511.
- ↑ Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population (англ.) // American Journal of Human Genetics (англ.)русск. : journal. — 2008. — August (vol. 83, no. 2). — P. 254—260. — DOI:10.1016/j.ajhg.2008.07.004. — PMID 18674747.
- ↑ «When Cells Stop Working» dated November 5, 2006 at Time Magazine
- ↑ Tanaka M., Nishigaki Y., Fuku N., Ibi T., Sahashi K., Koga Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases (англ.) : journal. — 2007. — DOI:10.1016/j.mito.2007.07.002. — PMID 17881297.
- ↑ Hello mothers, hello father (англ.). The Economist (27 October 2012). Дата обращения 17 декабря 2013.
Ссылки
На русском языке
На английском языке
Актуальные вопросы лечения митохондриальных нарушений uMEDp
Среди ярких событий современной медицинской науки одно из значимых мест занимает появление области, которую все чаще называют «митохондриальной медициной». Она интересна со многих точек зрения. Во-первых, как и полагается новому систематическому объединению, она знаменует собой выделение новых патологических процессов и нозологических форм. Во-вторых, ее безусловное прикладное значение определяется наличием специфической, так называемой энерготропной, терапии.
Первичные и вторичные митохондриальные нарушения
Ключевая область этого раздела медицины – наследственные синдромы, в основе которых лежат мутации генов, ответственных за митохондриальные белки (синдромы Кернса – Сейра, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), MERRF (myoclonic epilepsy with ragged red fibers), Пирсона, Барта и др.). Однако класс состояний, характеризующихся митохондриальной недостаточностью, отнюдь не ограничивается этими «первичными» митохондриальными заболеваниями. Огромное количество болезней включает в себя нарушения клеточного энергообмена в качестве «вторичных» звеньев патогенеза: синдром хронической усталости, мигрени, кардиомиопатии, гликогенозы, заболевания соединительной ткани, диабет, рахит, тубулопатии, панцитопения, гипопаратиреоз, печеночная недостаточность и др. Особое значение для практической медицины имеет изучение указанных нарушений в связи с разработкой в этой области эффективных методов терапевтической коррекции.
К настоящему времени наиболее изучены дефекты, связанные с дефицитом различных комплексов дыхательной цепи и некоторых ферментов матрикса. Появляются клинические описания дефекта и других ферментов, например, наружной митохондриальной мембраны [1], так что до полного представления о тонких механизмах митохондриальной дисфункции еще далеко, и часто речь идет о недостаточности митохондриальной функции в целом. Ткани и органы зависят от митохондриальной активности в различной степени [2–5]. В их ряду на первом месте стоят нервные элементы, затем сердечная и скелетная мышечная ткани, почки, эндокринные железы и печень. Увеличение количества митохондрий и их структурные нарушения широко определяются в эндотелиальных клетках, гладких миоцитах и перицитах различных сосудов [6].
К болезням, причиной которых являются мутации митохондриальных генов, относятся синдромы Кернса – Сейра (нарушения со стороны глаз, атаксия, мышечная слабость, нарушения сердечной проводимости и другие симптомы), Пирсона (вялость, нарушения со стороны крови, кишечника, поджелудочной железы), MELAS (энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды), оптическая нейропатия Лебера и многие другие. Причем описание таких синдромов продолжается и сейчас – так, в XXI веке уже опубликованы описания нескольких новых заболеваний.
Поскольку все митохондрии достаются новому организму только от цитоплазмы яйцеклетки, многие митохондриальные заболевания являются или спорадическими, или наследуются с нарушением законов Менделя – «внеядерное» или «цитоплазматическое» наследование по материнской линии. Распространенность этих болезней плохо изучена, но ясно, что они относятся к сравнительно редким наследственным заболеваниям. Отсюда и малый интерес широкого круга медиков к митохондриальной патологии.
Однако эти заболевания, хотя и создали ядро «митохондриальной медицины», отнюдь не составляют всего ее спектра. В последние годы внимание медиков начал все больше приковывать следующий факт: несмотря на наличие в митохондриях собственной ДНК, кодируются ею всего около 2% белков, используемых в митохондриях. Иными словами, 98% наследственной информации о митохондриальных белках заложено в ядре, а значит, количество наследственных митохондриальных нарушений, связанных с ядерными мутациями, должно быть несоизмеримо больше тех, о которых упоминалось выше. А известно их на сегодняшний день не так много (среди них различные формы младенческих миопатий, болезни Альперса, Лея, Барта, Менкеса, синдромы недостаточности карнитина, некоторых ферментов цикла Кребса и дыхательной цепи), что понятно – маленькую митохондриальную ДНК гораздо легче изучать, чем гигантский ядерный геном. Таким образом, значительное число таких состояний сейчас можно предсказать только гипотетически.
Интенсивное изучение признаков болезней клеточной энергетики приводит к еще более важному выводу: распространенность состояний, связанных с митохондриальной недостаточностью, не ограничивается наследственными синдромами, вызываемыми мутациями генов, непосредственно ответственных за митохондриальные белки. Умеренные нарушения клеточной энергетики могут не проявляться в виде самостоятельного заболевания, однако сказываются на характере течения других болезней. Широчайший круг других заболеваний включает в себя те или иные нарушения клеточной энергетики как вторичные звенья патогенеза.
Проведенные недавно в нашем институте исследования группы из 100 детей, поступивших в генетическую клинику с недифференцированными нарушениями физического и нервно-психического развития, показали, что у 49 из них отмечены нарушения клеточного энергообмена. Кроме того, нами выявлено влияние митохондриальной недостаточности на характер послеожогового рубцевания у детей, течение тонзиллитов, некоторых кардиологических, наследственных соединительнотканных, урологических и других заболеваний. Изучение этих патологических состояний и распространение информации о важности анализа энергетических дисфункций тем более актуально, что в настоящее время существуют действенные методы коррекции митохондриальной недостаточности, которые помогают в лечении перечисленных выше, не всегда истинно «митохондриальных» заболеваний.
Многие факторы окружающей среды и лекарственные препараты, вероятно, представляют собой существенную причину патологических изменений митохондрий. К этим факторам относится действие алкилирующих агентов (например, нитрозамины из окружающей среды), гидроксильных радикалов, высоких доз ультрафиолетового и ионизирующего излучений, лекарственных препаратов (бриостатин, азидотимидин), других химических агентов (аллоксан, цианиды, моноокись углерода и др.). Причиной митохондриального повреждения может быть и недостаточность некоторых микроэлементов, например селена. Во многих случаях чувствительность митохондриальной ДНК к действию повреждающих факторов окружающей среды в несколько раз выше по сравнению с чувствительностью ядерного генома. В целом область патогенетически целесообразного использования препаратов, воздействующих на митохондрии, включает в себя:
Лечение:
- митохондриальных болезней;
- «вторичных» (эндогенных и экзогенных) митохондриальных нарушений при других заболеваниях и состояниях.
- возможных осложнений различных заболеваний у пациентов с энергодефицитным диатезом;
- преждевременных патологических нарушений, связанных с пожилым возрастом.
- Реабилитационные мероприятия при различных хронических заболеваниях.
Кроме того, целесообразно применение энерготропных препаратов в качестве стимуляторов адаптационных процессов при заболеваниях, не несущих митохондриальной дисфункции в качестве патогенетической составляющей. Эта область наименее изучена, однако, по нашим данным, повышенная митохондриальная пролиферация может обладать важным адаптационным потенциалом, компенсирующим функциональный дефект при некоторых заболеваниях (например, при врожденных структурных миопатиях).
Общая характеристика энерготропных препаратов
Потенциальные возможности лечения митохондриальных болезней распределяются по трем основным направлениям [7]:
- Применение фармакологических препаратов и биологически активных добавок.
- Модификация макронутриентной поддержки, диетотерапия.
- Использование реабилитационных методов лечебной физкультуры.
Терапевтические подходы к лечению митохондриальных болезней подразделяются на семь категорий [8]:
- паллиативная терапия;
- удаление вредных метаболитов;
- применение искусственных акцепторов электронов;
- применение метаболитов и кофакторов;
- применение поглотителей кислородных радикалов;
- генная терапия;
- генетическое консультирование.
Большая группа лекарственных препаратов, которые принято называть метаболическими, пользуется чрезвычайной популярностью у широкого круга врачей. Можно без преувеличения сказать, что лекарства и нелекарственные средства, в разных количествах и соотношениях содержащие аминокислоты и пептиды, витамины и витаминоподобные вещества, коферменты и микроэлементы, применяются во всех областях медицины и по любому поводу. Такая популярность, очевидно, может объясняться как их эффективностью при лечении разнообразных патологических состояний, так и относительной безвредностью. Это сочетание факторов приводит к тому, что врачу легче назначить тот или иной препарат «на всякий случай», чем разбираться в целесообразности такого назначения. В результате из-за бездумного применения, из-за отсутствия методологической базы страдает эффективность лечения, что, в свою очередь, часто порождает сомнение в его принципиальной результативности. Все это диктует необходимость создания рациональной концепции применения лекарственных средств, относимых к метаболическим [9].
Важная группа таких препаратов представлена так называемыми энерготропными средствами, то есть средствами, усиливающими интенсивность обмена энергии на клеточном уровне. Наибольшее значение в контексте настоящей статьи имеют препараты (в таблице представлены некоторые из них), воздействующие на процессы, происходящие в универсальных клеточных органеллах – митохондриях. Митохондрии выполняют много функций, однако их основная задача – образование молекул аденозинтрифосфата (АТФ) в биохимических циклах клеточного дыхания. Накопленная энергия в последующем используется в других участках клетки.
Нарушения функций митохондрий относятся к важнейшим (часто ранним) этапам повреждения клеток. Эти нарушения ведут к недостаточности энергообеспечения клеток, к нарушению многих других важных обменных процессов, к дальнейшему развитию клеточного повреждения, вплоть до гибели клетки. Для клинициста оценка степени митохондриальной дисфункции имеет существенное значение как для формирования представлений о сути и степени происходящих на тканевом уровне процессов, так и для разработки плана терапевтической коррекции патологического состояния. Степень выраженности патологического процесса в том или ином органе связана со степенью зависимости его тканевых элементов от эффективности аэробного окисления.
Несмотря на то что в основе митохондриальных заболеваний могут быть сотни первичных биохимических дефектов, основные изученные звенья патогенеза, на которых и основаны современные подходы к коррекции митохондриальной недостаточности, связаны с нарушением реакций окисления пирувата до ацетил-КоА с помощью пируватдегидрогеназного комплекса; окисления ацетил-КоА до углекислого газа и образования восстановленных носителей электронов NADH и FADh3; реокисления восстановленного коэнзима Q ферментами электронно-транспортной цепи внутренней митохондриальной мембраны; транспорта свободных жирных кислот через мембрану митохондрии в виде эфиров карнитина; окислительного дезаминирования аминокислот с последующим поступлением их углеродного скелета в цикл Кребса; перекисного окисления и образования свободных радикалов.
Оценка достоверной эффективности энерготропных препаратов при митохондриальных болезнях сложна по многим причинам [7]. Вариабельность комплексных фенотипов затрудняет сравнение даже двух отдельно взятых больных с одним и тем же заболеванием. Поражение различных органов усложняет сравнительную оценку эффективности результата в целом. Большой проблемой является отсутствие четких критериев оценки динамики заболевания, наиболее выраженные признаки которого – это такие спорадические события, как инсультоподобные эпизоды или судороги.
Все это во многом объясняет тот факт, что, проанализировав в огромной работе 1335 источников о лечении митохондриальных болезней, Джералд Пфеффер (G. Pfeffer) и соавт. [7] смогли отобрать только 12 исследований, строго соответствующих критериям рандомизированного клинического исследования, причем в большинстве случаев они касались воздействия на нервно-мышечные проявления митохондриальных заболеваний с возможностью долговременной оценки таких признаков, как мышечная сила.
Проблема выбора дозы энерготропных препаратов
Сложность проблемы определяется, в частности, двумя факторами: во-первых, бытующим требовательным ожиданием заместительного эффекта при терапии митохондриальных болезней, а во-вторых, недоверием многих клинических биохимиков и фармакологов к возможности легкого введения тех или иных органических молекул внутрь митохондрии. Исходя исключительно из подобных теоретических соображений, ставятся, например, под сомнение как обоснованность применения янтарной кислоты, одного из ключевых метаболитов митохондрий, так и достоверность наблюдаемых позитивных эффектов этого препарата. Однако недавними исследованиями М.Н. Кондрашовой и сотрудников ее школы показано, что терапевтический эффект янтарной кислоты основан не на заместительном принципе, а на сигнальном. Таким образом, чтобы получить эффект, совершенно не нужно заполнять все митохондрии во всех клетках организма янтарной кислотой путем искусственного введения в больших количествах, достаточно назначить микродозы (5–10 мг/кг/сут). В нашей работе, используя новые диагностические приемы с применением транскутанного мониторирования рО2 и рСО2, мы выявили наличие подобного эффекта у L-карнитина. Вполне вероятно, что подобный же принцип может быть применен и к другим лекарственным веществам, используемым в терапии полисистемных нарушений цитоэнергетики.
Кроме того, в настоящий момент недоверие к возможности введения тех или иных молекул в митохондрию значительно поколеблено благодаря открытию большого и сложного комплекса транспортных систем, обслуживающих эти органеллы [10]. Нельзя не упомянуть о работах, показывающих эффективность применения энерготропных препаратов в достаточно высоких дозах. Так, в работе Р. Боулса [11] описана эффективность применения больших доз коэнзима Q10 (10 мг/кг/сут, но не более 200 мг в сутки) и L-карнитина (100 мг/кг/сут, но не более 2 г в сутки) в лечении синдрома циклической рвоты и других состояний, предположительно связанных с митохондриальными дисфункциями, – мигренозной головной боли, миалгии и синдрома множественных локальных болей.
В настоящее время нет единого понимания, какая длительность курса может быть оптимальной при энерготропной терапии. Естественно, во многих случаях (например, при лечении хронических заболеваний) необходимо достаточно длительное лечение, особенно если принимать во внимание вероятность заместительного механизма действия. Однако, исходя из практического опыта многих клиницистов и с учетом рекомендаций патофизиологов, длительное постоянное применение энерготропных препаратов (во всяком случае некоторых) не нужно. Целесообразнее применять схемы с периодическими назначением (1–3 месяца) и отменой (примерно на такой же или несколько больший период). Таким образом, совершенно актуально использование как высокодозовых и длительных, так и низкодозовых кратковременных схем применения энерготропных препаратов. Поскольку выявление сигнальных и заместительных составляющих эффекта для большинства энерготропных препаратов – дело будущего, выбор схемы применения до сих пор зависит от искусства врача.
Коэнзим Q10
Коэнзим Q (коэнзим Q10, убихинон, витамин Q10) – небольшая жирорастворимая молекула, непосредственно участвующая в транспорте электронов по дыхательной цепи митохондрий. Она свободно диффундирует в мембранном бислое и помимо электронов передает ферментному комплексу III также и протоны, которые захватывает из водной среды. Убихинону свойственны витаминоподобные функции. Будучи введенным в организм, он оказывает значительный антиоксидантный эффект, повышает продукцию АТФ и стабилизирует состояние кальциевых каналов.
Коэнзим Q – один из наиболее распространенных и эффективных энерготропных препаратов [12, 13]. В суточных дозах 300–1500 мг он эффективен при дефиците убихинона, дефектах второго и третьего ферментных комплексов дыхательной цепи, клинически выражающихся в синдроме MILS (maternally inherited Leigh syndrome), синдромах MERRF, MELAS и Кернса – Сейра. Его высокая клиническая эффективность в лечении атаксии Фридрейха и других нейродегенеративных заболеваний показана в нескольких работах [14].
Эффект высоких доз коэнзима Q10 (600 мг 2 раза в день перорально в течение двух месяцев) у больных с синдромом MELAS, прогрессирующей наружной офтальмоплегией и некоторыми другими формами митохондриальных болезней был изучен в двойном слепом плацебоконтролируемом исследовании [7, 15]. Было показано, что на фоне увеличения концентраций коэнзима Q10 в крови снижался уровень лактата при кратковременной, но не при долгосрочной нагрузке. Достоверных изменений других биохимических показателей отмечено не было.
Так же как и для многих других энерготропных препаратов, примеры эффективного применения коэнзима Q10 часто можно обнаружить в работах, посвященных терапии состояний, связанных с различными вторичными проявлениями тканевой гипоксии. Так, Д.М. Ароновым и соавт. [16] было проведено рандомизированное проспективное исследование коэнзима Q10 (препарат Кудевита®) в лечении пациентов с ишемической болезнью сердца с сердечной недостаточностью II–III функционального класса. Пациенты основной группы принимали препарат в дозе 150 мг/сут: по 2 капсулы (60 мг) утром и 3 капсулы (90 мг) вечером. Одновременно пациенты основной и контрольной групп получали стандартную терапию, показанную при данном заболевании. В процессе исследования не назначали препараты, влияющие на метаболизм миокарда, кардиопротекторы и антиоксиданты (триметазидин, мельдоний, оксиметилэтилпиридина сукцинат, инозин, аденозинтрифосфат, кокарбоксилаза, витаминные и иные метаболические средства). Длительность наблюдения составила 3 месяца.
В результате было показано, что лечение больных хронической сердечной недостаточностью на фоне ишемической болезни сердца и перенесенного инфаркта миокарда, находящихся на поликлиническом наблюдении, оказало за относительно небольшой отрезок времени существенное влияние на состояние сердечно-сосудистой системы. При этом препарат способствовал снижению диастолического артериального давления, достоверному улучшению физической работоспособности, сократительной функции сердца и гемодинамики по данным ультразвукового исследования (УЗИ), улучшению картины электрокардиограммы (ЭКГ), указывающей на положительные сдвиги в метаболизме миокарда, липидного профиля плазмы крови. Клинически отмечалось уменьшение количества и выраженности приступов боли в груди; уменьшалась частота приема нитроглицерина.
Препараты, содержащие коэнзим Q10, хорошо знакомы врачам разных специальностей, но до недавнего времени они не могли широко применяться в лечебной практике, так как были представлены в России в виде БАД. Кудевита® в настоящее время является единственным зарегистрированным в России безрецептурным лекарственным препаратом с активным действующим веществом убидекаренон (коэнзим Q10) и, несомненно, поможет более успешно лечить различные заболевания у детей.
L-карнитин
Карнитин – низкомолекулярное соединение, производное аминомасляной кислоты. В тканях млекопитающих присутствует только L-стереоизомер (левокарнитин), именно он биологически эффективен. Карнитин принимает непосредственное участие в катаболизме липидов, обеспечивая перенос длинноцепочечных жирных кислот в виде сложных эфиров (ацилкарнитинов) из цитоплазмы через наружную и внутреннюю митохондриальные мембраны в матрикс митохондрий. Внутри митохондрий транспортированные жирные кислоты подвергаются бета-окислению с образованием ацетил-КоА, который служит субстратом для цикла трикарбоновых кислот Кребса и последующего синтеза АТФ в организме. Наряду с этим окисление жирных кислот – главный путь кетогенеза, а кетоновые тела являются дополнительным энергетическим источником для периферических тканей и головного мозга.
Влияние карнитина на жировой обмен осуществляется также его участием в цитоплазматическом синтезе жирных кислот путем обратного переноса необходимых для этого ацетильных групп митохондриального ацетил-КоА через митохондриальную мембрану в цитоплазму. Помимо перечисленного, карнитин регулирует отношение «ацил-КоА/свободный КоА» в митохондриях. Связывая ацильный радикал, он высвобождает КоА и тем самым активирует интенсивность энергетического метаболизма в тканях. Исключительное значение карнитина становится очевидным в условиях высокого расходования энергетических ресурсов – при заболеваниях, усиленных физических или эмоциональных нагрузках, а также при недостаточном питании. После истощения запасов углеводов липиды становятся главным источником синтеза АТФ в организме.
Другая важная функция карнитина заключается в его способности образовывать соединения с различными органическими кислотами, являющимися промежуточными продуктами окислительных процессов. Данные вещества, накапливаясь в митохондриях и цитоплазме клеток, оказывают мембранотоксическое действие и ингибируют активность ряда ферментов. Выведение этих токсичных органических соединений из организма происходит через почки в виде ацилкарнитинов.
Левокарнитин высокоэффективен при лечении как первичных форм дефицита карнитина, так и широкого круга заболеваний, связанных со вторичным снижением его содержания в организме. Кроме того, занимая уникальное положение относительно митохондрии и стимулируя приток в нее энергосубстратов, карнитин является универсальным стимулятором тканевого энергообмена, что актуально не только в отношении компенсации энергодефицита, но и в отношении компенсаторной адаптации практически к любым структурно-функциональным дефектам. Данные об эффективности применения препаратов L-карнитина у больных митохондриальными заболеваниями достаточно многочисленны [13, 17], хотя чаще описывают их применение в комплексе с другими энерготропными средствами (см. ниже).
Отдельно следует упомянуть о важности применения L-карнитина при формах его наследственной недостаточности. В качестве примера упомянем работу Е.А. Николаевой и соавт. [18], в которой была показана эффективность применения препарата Элькар® в дозе 800 мг/сут с двухлетнего возраста у больного с первичным системным дефицитом карнитина. Динамическое наблюдение в течение 2 лет показало выраженное улучшение самочувствия и состояния мальчика на фоне терапии Элькаром. Ребенок и его родители не предъявляли жалоб, связанных с лечением препаратом, физическое развитие в возрасте 4 лет было средним (вырос на 19 см), мышечный тонус физиологическим. В результате лечения отмечены положительные изменения со стороны сердца, улучшились биохимические показатели крови.
Креатин
Моногидрат креатина представляет собой дополнительный источник энергии. В отличие от нестабильных форм – чистого креатина и фосфата креатина, – моногидрат креатина прекрасно всасывается и с успехом применяется в спортивной медицине. Отмечена его эффективность в лечении различных митохондриальных болезней: синдромов Лея, Кернса – Сейра, MELAS и др. [14, 19–21]. Однако существуют и противоположные данные, не подтверждающие его эффективность при митохондриальных болезнях [12].
В рандомизированном плацебоконтролируемом исследовании [7, 22, 23] эффективности моногидрата креатина, применяемого в течение трех недель (4–10 г в сутки) у больных синдромом MELAS и митохондриальной миопатией, отмечено достоверное увеличение мышечной силы и снижение уровня лактата после нагрузки. В двойных слепых плацебоконтролируемых исследованиях у больных с прогрессирующей наружной офтальмоплегией и митохондриальной миопатией [24] (20 г в сутки в течение месяца) и у больных с хронической прогрессирующей наружной офтальмоплегией и с синдромом Кернса – Сейра [25] (150 мг/кг в течение 6 недель) клиническая эффективность моногидрата креатина не выявлена.
Дихлорацетат
Дихлорацетат – активатор пируватдегидрогеназы – уже значительное время активно изучается в качестве возможного средства исправления митохондриальных функций [12]. В ряде работ показана его эффективность при лечении синдрома MELAS [26, 27], дефицита пируватдегидрогеназного комплекса [28], а также при исправлении митохондриальных функций в опухолевых клетках, ведущем к позитивному эффекту при лечении рака. Некоторые исследователи столь высоко оценивают его лечебный потенциал, что назначают этот препарат несмотря на то, что применение дихлорацетата (в частности в суточных дозах 25 мг/кг) может вызывать развитие периферических полиневропатий [29].
В двойном слепом плацебоконтролируемом исследовании эффективности дихлорацетата у больных с различными митохондриальными заболеваниями [7, 30] (50 мг/кг в сутки в течение недели двумя курсами с интервалом три месяца) было отмечено значительное снижение концентраций лактата, пирувата и аланина в крови (в покое и после нагрузки), а в мозге – значительное снижение соотношения «лактат/креатин» и повышение соотношений «холин/креатин» и «ацетиласпартат/креатин». В других подобных работах [28, 31, 32] была также отмечена нормализация уровня лактата (а в первых двух из этих работ – и пирувата) после приема дихлорацетата.
Янтарная кислота
Сукцинат – один из эффективных медиаторов транспорта электронов, успешно использующийся при острых нарушениях тканевого дыхания. В отношении хронических расстройств целесообразность его применения спорна, однако в научной литературе приводятся описания эффективности сукцината при недостаточности I дыхательного комплекса [33] и при синдроме MELAS (длительная монотерапия суточной дозой 6 г) [34].
Фолиевая кислота
Фолиевая кислота – водорастворимый витамин (В9), необходимый в первую очередь при активной репликации ДНК, то есть делении клеток. Однако описан выраженный положительный эффект этого витамина в суточной дозе 1–2,5 мг/кг при синдроме Кернса – Сейра [33].
L-аргинин
L-аргинин – незаменимая для детей аминокислота, снабжающая азотом систему NO-синтаз. В настоящее время растет число указаний [12] на эффективность применения L-аргинина при синдроме MELAS, в частности, в отношении терапии инсультоподобных эпизодов и сердечно-сосудистых нарушений. Так, например, с использованием позитронно-эмиссионной томографии показано, что применение L-аргинина эффективно при лечении кардиомиопатии при синдроме MELAS [35].
Другие вещества
Наряду с вышеперечисленными, к веществам, несомненно позитивно влияющим на клеточный энергообмен, относят витамин Е (альфа-токоферол), витамин С (аскорбиновая кислота), липоевую кислоту, глутатион, рибофлавин, тиамин и др. Однако в литературе, посвященной митохондриальным болезням, пока не находится четких доказательств их эффективности (по крайней мере, в моноварианте), хотя эти вещества часто используются в комплексных схемах энерготропной терапии.
В настоящее время в различных, в первую очередь экспериментальных, работах активно исследуются новые вещества, представляющие собой потенциально перспективные препараты для лечения митохондриальных заболеваний – антиоксиданты в соединении с трифенилфосфониевым катионом (митохинон, MitoVitE, MitoTEMPOL, MitoPBN, смесь Скулачева), тролокс, SS-пептиды (Szeto-Schiller peptides) [14, 36, 37], ресвератрол [38], препараты, влияющие на сборку дыхательных комплексов [39], оптимизирующие обмен кальция [40], активаторы митохондриального биогенеза [41] и др.
Комплексная энерготропная терапия
Спектр потенциальных патологических нарушений клеточного энергообмена чрезвычайно велик (повреждения различных звеньев цикла Кребса, дыхательной цепи, бета-окисления и др.). Хотя перечень энерготропных препаратов также достаточно широк, далеко не всегда имеется возможность выявить конкретное точечное повреждение митохондрий и точно подобрать подходящий лекарственный препарат. В связи с этим наиболее эффективными в широкой клинической практике могут быть комплексы энерготропных препаратов, обладающих способностью воздействовать сразу на несколько ключевых этапов клеточного энергообмена. При этом на первое место по значимости выдвигаются такие препараты, как L-карнитин, коэнзим Q10, цитохром С и их комплексы с другими вышеперечисленными лекарственными средствами [42–44]. Схемы лекарственной коррекции цитоэнергетической недостаточности у детей активно разрабатываются в настоящее время в Московском НИИ педиатрии и детской хирургии и в Российском национальном исследовательском медицинском университете им. Н.И. Пирогова.
Так, данные Е.А. Николаевой свидетельствуют о том, что при митохондриальных энцефаломиопатиях комплексная энерготропная терапия позволяет добиться существенного клинического эффекта во всех сферах проявления патологического процесса. Результатом лечения является нарастание массы тела, уменьшение выраженности сердечно-сосудистых нарушений, снижение частоты приступов рвоты, судорог, уменьшение выраженности проявлений энцефалопатии и миопатии, снижение утомляемости. Некоторые примеры свидетельствуют о том, что эффективность правильно подобранной энерготропной терапии даже при тяжелых «первичных» митохондриальных синдромах может быть поразительной. В качестве одного из примеров можно привести историю болезни ребенка с синдромом Барта – одним из таких синдромов, клиническая картина которого характеризуется задержкой роста и психомоторного развития, миопатией, кардиомиопатией, нарушениями со стороны крови. Многолетнее лечение комплексом препаратов, включавшим в себя коэнзим Q10, цитохром С, L-карнитин (Элькар) и некоторые другие, привело к тому, что в подростковом возрасте главной проблемой этого мальчика с тяжелым наследственным заболеванием стали попытки избежать постановки на воинский учет.
В двойном слепом плацебоконтролируемом исследовании [7, 14, 45] у больных с такими митохондриальными заболеваниями, как синдром Кернса – Сейра, синдром MELAS, хроническая наружная офтальмоплегия, оптическая нейропатия Лебера, митохондриальная нейрогастроинтестинальная энцефаломиопатия, а также с митохондриальными заболеваниями с редкими точковыми мутациями была проведена оценка двухмесячного комплексного применения креатина, коэнзима Q10 и липоевой кислоты. В этой работе выявлено статистически достоверное снижение уровней лактата в плазме крови и 8-изопростана – в моче. У больных с синдромом MELAS отмечено также нарастание массы тела (не за счет жировой ткани). Есть указания [46] на то, что в лечении митохондриальных болезней (как психических, так и соматических нарушений) могут быть эффективны комплексы, содержащие коэнзим Q10 (200–400 мг/сут) и рибофлавин (100–400 мг/сут), в некоторых случаях с добавлением витамина С (1000 мг/сут), витамина Е (400 МЕ/сут), карнитина (2000 мг/сут), креатина (5000 мг/сут) и магнезии (250–500 мг/сут).
При заболеваниях, включающих в свой симптомокомплекс «вторичную» митохондриальную недостаточность, также можно добиться улучшения качества жизни больных. Приведем весьма «эффектный» пример: у низкорослых детей c различными неэндокринными наследственными заболеваниями на фоне лечения энерготропными препаратами – L-карнитином (Элькар), коэнзимом Q10 и другими – удается достичь значительной стимуляции роста – до 6–7 см в год. При некоторых заболеваниях благодаря энерготропной терапии впервые была продемонстрирована возможность относительного успеха в лечении (например, при лечении синдрома Ретта и туберозного склероза отмечено улучшение когнитивных и эмоциональных функций). Существенный позитивный эффект применения энерготропных препаратов наблюдался и в ряде других отделений нашего института: урологическом (при комплексном лечении гидронефроза и гиперактивного мочевого пузыря), ожоговом центре (при реабилитации детей после ожогов), кардиологии (при лечении кардиомиопатий, миокардиодистрофии и нарушений сердечного ритма), пульмонологии (при лечении ряда хронических заболеваний легких) и др.
Применение средств метаболической коррекции позволило оказать существенное влияние на состояние здоровья детей дошкольного возраста с различными вариантами нарушения речевого развития (общее недоразвитие речи, дислалия, задержка психоречевого развития), у детей с соединительнотканной дисплазией и в группе так называемых часто болеющих детей. Эти работы выполнялись нами совместно со специалистами Российского государственного медицинского университета им. Н.И. Пирогова (группа С.О. Ключникова). В лечении этих групп детей были применены комплексы, включающие коэнзим Q10, L-карнитин (Элькар), ряд других энерготропных препаратов. Указанное лечение дети получали, как правило, длительно, в течение 2–3 месяцев, после чего они проходили повторное обследование, позволившее выявить существенную положительную динамику в состоянии здоровья. Отмечались минимизация предъявляемых жалоб, улучшение сна и аппетита, исчезновение или снижение выраженности ряда клинических признаков заболеваний, нормализация лабораторных показателей; возрастала выносливость в отношении физических и интеллектуальных нагрузок.
Заключение
Все вышесказанное свидетельствует о необходимости научно-прикладных разработок, направленных на создание современных принципов энерготропного лечения (по отработке состава энерготропных комплексов, тщательному подбору доз активных веществ, определению оптимальных схем назначения, в том числе с учетом хронобиологических ритмов). Приведенные выше примеры свидетельствуют о необходимости именно комплексного использования таких средств. Однако при каждой нозологической форме должны разрабатываться свои специализированные комплексы, включающие патогенетически наиболее значимые компоненты клеточного энергообмена (например, коэнзим Q10, L-карнитин, цитохром С, янтарная кислота и др.).
Что такое митохондриальные синдромы
Мелас-синдром – это нейродегенеративное заболевание, которое характеризуется прогрессирующим течением.
Что такое митохондрия? Эта органелла является энергетическим источником клетки. Она ответственна за производство АТФ, которая дает возможность синтезировать белки, жиры и углеводы. Располагается она на внутренней оболочке клетки и тесно с ней соприкасается. Энергия появляется при захвате молекулы глюкозы и ее превращения внутри органеллы в пируват.
Митохондрия — это потомок бактерий. Поэтому в ней имеется своя кольцевая хромосома и несколько функционирующих генов. При их мутации появляются митохондриальные заболевания. Выделяют две группы болезней, связанных с этими органеллами. Наследственные болезни с мутацией генов, ответственных за митохондриальные белки;
- Синдром Барта;
- Пирсона;
- Кернса-Сейра;
- MERRF;
- MELAS.
И вторичные митохондриальные заболевания.
Наследование этих болезней происходит по материнской линии. При этом дефектный ген передается обоим потомкам. Ведь яйцеклетка снабжает новый организм цитоплазмой и митохондриями. В то время как, сперматозоид вносит в новый организм только половину генома.
Митохондриальные синдромы
Под этим термином понимают нарушения, которые происходят при нарушении работы энергетической системы клетки. Они обусловлены мутацией митохондриальной ДНК, его истощением и нарушением в структуре ядерной дезоксирибонуклеиновой кислоты.
Синдромы делеции митохондриальной ДНК
Сокращенно его называют СДМ или MSD. Эта патология передается менделевским наследованием. Симптомы недуга появляются после рождения. Клиника – мышечная слабость, проблемы с печенью, редко появляются аномалии ЦНС. У младенца преобладает гипотонус, он плохо питается и усваивает пищу, отстает в развитии. Реже выражены приступы эпилепсии и офтальмоплегия. Встречается кардиомиопатия с судорогами. Прогноз при заболевании неблагоприятный.
Точечные мутации мт ДНК
Точечные мутации в митохондриальной ДНК представлены заменой оснований, делециями и инсерциями. Они возникают как спорадически, так и наследуются по материнской линии. Точечные мутации ассоциированы с множественными патологическими процессами в органах и тканях (ЦНС, почки, печень, эндокринная система, сердце и мышцы).
Точечные мутации лежат в основе синдрома MELAS. Поражаются транспортная РНК и ее гены. При этом в 90% случаев это дефект A3243G в митохондриальном гене MTTL1. Среди населения Австралии ее носителем являются 236 человек на 100000 населения. Выявлено, что эта мутация является фактором риска развития инсультоподобных эпизодов. А ее носители чаще подвержены сердечно-сосудистым заболеваниям.
Синдром истощения митохондриальной ДНК
Это синдром, который формируется антенатально. Первые признаки можно заметить еще до рождения малыша. Это гидронефроз почек, субэпенимальные кисты головного мозга. В первые сутки ребенок угнетен, отмечается гипотония мышц, состояние, похожее на сепсис. Прогноз при болезни неблагоприятный. Смерть наступает в течение первой недели от полиорганной недостаточности или сопутствующих инфекций.
Синдром обусловлен мутацией в гене FBXL4. Она происходит в результате нарушения репликации митохондриальной ДНК. В результате снижается активность комплексов дыхательной цепи митохондрий и появляются разнообразные нарушения во многих системах и органов.
Синдром NARP
Это редкое орфанное заболевание, которое дословно расшифровывается как нефропатия, атаксия и пигментный ретинит. В мкб 10 заболевание получило шифр G31.8. Основные клинические проявления болезни: сенсо-моторная нейропатия, мозжечковая атаксия и куриная слепота.
Распространённость заболевания – 8 заболевших на 100000 населения. Болезнь передается по материнской линии, путем мутации в митохондриальной ДНК.
Клиническая картина развивается при нарушении картины окислительного фосфорелирования. При этом нарушается синтез энергии в клетки. Особенно от этого страдают ткани, зависимые которые имеют массивные энергозатраты (мышцы и нервная ткань).
Митохондриальная энцефалопатия с лактат-ацидозом и инсультоподобными эпизодами
Митохондриальная энцефалопатия — это нейродегенеративный процесс. При нем нарушается работа многочисленных систем и органов. Развивается диабет, падает слух, страдает психика. Пациенты не могут переносить физические нагрузки.
Причины болезни: точечные мутации в генах митохонриальной ДНК. Наследование проявляется только по материнской линии. Вероятные носители могут иметь бессимптомное течение болезни. Тогда их можно определить только по результатам биопсии мышц. Остальные носители имеют одно из проявлений болезни MELAS.
Важно! Известно около 10 генов, которые участвуют в патогенезе заболевания.
Полная клиническая картина болезни:
- — инсультоподобные эпизоды;
- — клиника появляется до 40 лет;
- — энцефалопатия с судорогами и деменциями;
- — страдание мышечных волокон;
- — лактат-ацидоз;
- — головная боль, переходящая в мигрень;
- — непереносимость физических нагрузок.
МКБ 10
Как это заболевание кодируется в международной классификации болезни.
| E 88.4 | Это мелас-синдром; |
| G 71.3 | Неклассифицированная мышечная миопатия. |
Причины
Наследование проходит по материнской линии. При наличии патологического гена у матери его получают все ее дети. Возможны варианты бессимптомного течения болезни. В этом случае носителя можно определить по результатам биопсии мышечной ткани.
К сожалению, возможны варианты спонтанных мутаций. В этом случае ген изменяется у пациента. Причиной могут стать неблагоприятные факторы внешней среды: радиация, воздействие химикатов, травмы.
Этиопатогенез митохондриальной патологии
В зависимости от нарушения патогенеза рассматривают 4 группы нарушений:
- Нарушение цикла Кребса;
- Накопление пирувата;
- Дефект фосфорелирования;
- Проблемы обмена жирных кислот.
В любом случае клетка прекращает свою нормальную работу. В тканях и органах не идут процессы развития и обновления. Однако, больше всего страдает мышечная и нервная ткань. Именно она отличается большой энергозависимостью.
Генетические изменения при митохондриальных заболеваниях
Митохондриальная генетика значительно отличается от менделевских законов. Материнское наследование идет ко всем рожденным детям. Ведь цитоплазму с органеллами будущий организм получает от яйцеклеток.
Часто в организме у больного встречается два типа мт ДНК. В одних нормальная или дикая ДНК, а в других патологическая мутантная. Такое явление носит название гетероплазмия. Оба типа ДНК случайным образом распределяются среди дочерних клеток. Поэтому предсказать развитие болезни крайне сложно.
Продолжительность жизни
К сожалению, патология является фатальной для пациента. Лечения не разработано. Продолжительность жизни является непредсказуемой. Особенно опасно, когда при синдроме сочетаются несколько клинических проявлений болезни. Смерть наступает в результате фатальных нарушений (инсульта, эпилепсии).
В детском возрасте заболевание приводит младенческой смертности. Ребенок плохо развивается и растет. Пища слабо усваивается. Рефлексы отсутствуют.
Частота встречаемости
Митохондриальная патология является редким заболеванием. Однако, в некоторых популяциях болезнь появляется достаточно часто. Например, синдром мелас выявили у 8 родившихся детей на 100 000 населения в общей популяции. А в Австралии с митохондриальным синдромом рождаются 238 детей на 100000 населения. Закрытое общество предрасполагает к распространению патологических генов.
Встречаемость у взрослых довольно сложно оценить. Ведь митохондриальная болезнь тяжело диагностировать. Кроме того, даже пациенты с патологическим генотипом могут являться «здоровыми» из-за преобладания «дикой» ДНК в генотипе.
Важно! В ограниченной популяции, например, в Финляндии, количество лиц с мутацией A32433G составляет 10 на 100000 населения.
Клиническая картина
Заболевание может проявляться в зрелом и детском возрасте. Его выраженность зависит от количества копий мутированного мт ДНК. Как правило, первые симптомы заметны в возрасте 5-6 лет. Болезнь протекает тяжело. Ребенок плохо переносит физические нагрузки. У него возникают мигнеподобные головные боли. Они сопровождаются приступами тошноты, рвоты. Во время приступа больной может терять сознание.
Опасные состояния, которые требуют реанимационных мероприятий – лактат-ацидоз в крови, инсультоподобные эпизоды. Приступы сопровождаются частичной утратой зрения, слуха, атаксией, невозможностью разговаривать. После припадка может случиться частичный паралич или парез.
При проведении МРТ или КТ изменения головного мозга свидетельствуют о перенесенном инсульте. Нарастают деменция и умственная недостаточность. Могут появляться судороги и эпилептические припадки.
Течение заболевание в зрелом возрасте более спокойное. Как правило, позднее проявление свидетельствует о большем количестве копий дикой мт ДНК. Они позволяли длительное время обеспечивать зависимые ткани энергией. Но при наступлении декомпенсации у пациента развивается аналогичная клиническая картина:
- Судороги;
- Мигрень;
- Острое нарушение мозгового кровообращения;
- Накопление лактата и пирувата в крови.
Со временем у пациента нарастают явления энцефалопатии и деменции. Они формируются на фоне прогрессирующих изменений нервных клеток и их гибели.
Диагностические критерии синдрома
- — Болезнь наследуется по материнской линии;
- — Первые симптомы проявляются в детском возрасте, однако, не позже 40 лет;
- — До появления первых признаков заболевания ребенок здоров;
- — Плохая переносимость физических нагрузок;
- — При биопсии мышц визуализируются рваные красные волокна;
- — Появляются приступы головных болей, похожих на мигрень. Они сопровождаются рвотой и тошнотой;
- — Лактат-ацидоз в крови;
- — Эпизоды, похожие на инсульт;
- — Прогрессирующая энцефалопатия с приступами эпилепсии и нарастающей деменцией;
- — Болезнь прогрессирует.
Заболевание стоит дифференцировать с другими митохондриальными патологиями, эпилептическим статусом, вирусным энцефалитом, васкулитом, коровьим бешенством и другими прионными болезнями, редкими видами инсультов.
Диагностические мероприятия
Диагностика синдрома Melas основана на определении высокого уровня лактата и пирувата в крови после физических нагрузок. Характерные изменения можно обнаружить в геноме пациента. Для этого проводится дорогостоящая лабораторная диагностика.
Нейровизуализация показывает вторичные изменения в головном мозге и внутренних органах. Но она не дает типичной картины заболевания. Важной задачей ученых и врачей является предотвращение рождения пациентов с этой патологией. Для этого при подозрении на заболевание или при наличии мутации у родственников проводят пренатальный скрининг.
КТ
На компьютерной томографии можно обнаружить косвенные признаки перенесенного инсульта. Очаги схожи с ишемическим проявлением болезни. Но они не локализуются в бассейне определенной артерии. Наиболее часто их обнаруживают в теменной, височной и затылочной долях.
Реже страдает лобная область, мозжечок и подкорковые структуры (базальные ганглии). При этом во время исследования можно обнаружить «свежие» и «старые» поражения. При этом приступы способны рецидивировать каждые 1-3 месяца.
МРТ
Инсульты при этой болезни возникают в молодом возрасте. Им предшествуют эпизоды длительных головных болей. На МРТ повреждение тканей не совпадают с бассейнами крупных артерий. Локализация очага — это кора, реже поверхностно повреждается белое вещество. И еще характерный МРТ признак — это его флуктуация. Зона поражения может исчезать и снова появляться. Меняются даже контуры «некроза».
Тесты для диагностики
Определить изменения в генах мт ДНК поможет генетическая лаборатория. Частые мутации – это замена A3243G, которая встречается в 80% случаев. Второй распространенной аномалией генов является дефект G13513A. Где можно пройти диагностику? Не каждая генетическая лаборатория выполняет этот анализ. В Москве тестирование доступно в МГНЦ РАМН (исследование наследственных болезней обмена веществ).
Второй тест – это биопсия мышечных волокон. Его проводят в Московском научно-исследовательском институте педиатрии и детской хирургии. Он актуален в том случае, если генетические тесты не выявили распространенных видов мутаций в геноме.
Пренатальная диагностика
Этот вид тестов проводят в случае установленных случаев заболевания в семье. Для этого используют инвазивный метод амниоцентез. Это забор амниотической жидкости во время беременности и обнаружение в ней лактата-пирувата. Исследование выполняют в стационаре под контролем УЗИ. Этот вид тестирования эффективен и для диагностики других митохондриальных патологий.
Лечение
Терапия заболевания направлена на купирование симптомов и улучшение энергетических процессов в клетке. В первом случае важен контроль за приступами эпилепсии. Ведь в результате могут развиваться инсультоподобные изменения. Но использование некоторых синдромальных препаратов (вальпроевой кислоты) органичено из-за негативного влияния на митохондрии.
Строение митохондрииПатогенетическое лечение только разрабатывается. Оптимальных способов восстановить энергетический обмен нет. Пациентам назначают коэнзим Q, антиоксиданты, идебенон, янтарную кислоту, цитохром С и аскорбинку. Но действие такой терапии крайне низкое.
Важно! У больных следует исключить использование препаратов, ингибирующих энергетический обмен. Это вальпроевая кислота, глюкокортикостероиды, хлорамфеникол, барбитураты.
Диета
При этом синдроме ограничивают потребление углеводов. Рекомендуемая доза до 10 г/кг в пище. Высокое содержание глюкозы в крови будет способствовать усилению проблем с энергетическим метаболизмом.
У пациентов отмечается нестойкий положительный эффект при переходе на кетогенную диету. В этом случае разрешено употреблять преимущественно белковые продукты и жиры. Типичный прием пищи — это стейк и немного спаржи.
Нейродиетология при митохондриальной патологии
Нейродиетология является наукой, которая находится на стыке двух учений: нейрофизиологии и диетологии. Она разрабатывает принципы питания при метаболических заболеваниях.
При синдроме Melas рекомендовано исключить из потребления те вещества, которые оказывают негативное влияние на метаболизм клетки. При болезни употребление глюкозы формирует «блок» в энергоцепи. Этот диетический принцип называется «обхождение блока».
Что рекомендуют ограничить? Некоторые жиры, сахар, алкоголь, глютен, крахмал, белок, кофеин, глутамат натрия. Следует избегать голодания, переедания и низкого количества энергии в пище. Дополнительно в рацион вводят добавки коэнзима Q10, L-карнитина, витамины группы B, никотинамид, биотин, альфа-липоевая кислота, фолиевая и янтарная кислота, селен, витамин К. Такая диета дает возможность длительно избегать обострений болезни.
Синдром Melas — это редкая наследственная патология, которую еще не научились лечить. Но врачи способны провести анализ на ранних сроках беременности и предотвратить рождение больного ребенка. А у пациентов остается надежда, что тайны митохондрий вскоре будут раскрыты.
Ольга Гладкая
Автор статей: практикующий врач Гладкая Ольга. В 2010 году окончила Белорусский Государственный Медицинский Университет по специальности лечебное дело. 2013-2014 – курсы усовершенствования «Ведение пациентов с хронической болью в спине». Ведет амбулаторный прием пациентов с неврологической и хирургической патологией.
Митохондриальный сахарный диабет — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 21 марта 2017; проверки требуют 2 правки. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 21 марта 2017; проверки требуют 2 правки.Митохондриа́льный са́харный диабе́т — группа генетически обусловленных заболеваний, наследующихся исключительно по материнской линии, проявляющихся сочетанием клинической картины сахарного диабета и сопутствующих неврологических заболеваний: тугоухость, миопатии или неврологические нарушения[1].
Причина митохондриальных форм сахарного диабета — точечная мутация в паре нуклеотидов 3243. Сахарный диабет, вызванный данной мутацией, может дебютировать в возрастном периоде от окончания пубертатного периода до 85-ти лет, чаще проявляется в третьем — пятом десятилетиях жизни[1].
Предположительно в основе патогенеза недостаточная секреция инсулина бета-клетками поджелудочной железы при сохранённой (обычной) чувствительности рецепторов периферических тканей к его действию[1]. Та же мутация может стать причиной менее частой, но более тяжёлой митохондриальной энцефалопатии с лактатацидозом и инсультоподобными эпизодами — синдром MELAS, развивающийся в раннем детском возрасте, начинается появлением двусторонней тугоухости, затем проявляется сахарный диабет, а инсультоподобные приступы и энцефалопатия наблюдаются в третьем-четвёртом десятилетии жизни[1].
Гипергликемия в момент выявления заболевания обычно невелика, легко компенсируется диетотерапией, но в дальнейшем имеет тенденцию к прогрессированию[1].
На сегодняшний день специфического лечения митохондриального сахарного диабета не существует. Обычно необходимость в проведении инсулинотерапии возникает с момента выявления заболевания[1].
Заболевание носит пожизненный характер, однако при соблюдении диеты и адекватной симптоматической терапии прогноз положительный.
На сегодняшний день молекулярная этиология заболевания установлена — точечная мутация в паре нуклеотидов 3243 — при рождении ребёнка с заболеванием следует обратиться в медикогенетическую консультацию (МГК) для уточнения диагноза и прогноза планирования семьи.
- ↑ 1 2 3 4 5 6 Кураева Т.Л., Зильберман Л.И. Неиммунные формы сахарного диабета у детей // International jornal of endocrinology. — 2010. № 7 (31). С. 101-106.