| Недоразвитие мозолистого тела головного мозга (нарушение межполушарного взаимодействия)
Развитие головного мозга ребенка начинается внутриутробно и активно продолжается после рождения.
По исследованиям физиологов правое полушарие головного мозга – гуманитарное, образное, творческое – отвечает за тело, координацию движений, баланс, пространственное зрительное и кинестетическое восприятие.
Левое полушарие головного мозга – математическое, знаковое, речевое, логическое, аналитическое – отвечает за восприятие – слуховой информации, постановку целей и построений программ.
Единство мозга складывается из деятельности двух полушарий, тесно связанных между собой системой нервных волокон (мозолистое тело).
Мозолистое тело (межполушарные связи) находится между полушариями головного мозга в теменно-затылочной части и состоит из двухсот миллионов нервных волокон. Оно необходимо для координации работы мозга и передачи информации из одного полушария в другое.
Агенезия (нарушение, недоразвитие) мозолистого тела искажает познавательную деятельность детей. Если нарушается проводимость через мозолистое тело, то ведущее полушарие берет на себя большую нагрузку, а другое блокируется. Оба полушарие начинают работать без связи.
Нарушаются пространственная ориентация, баланс, осознание собственного тела, адекватное эмоциональное реагирование, координация работы зрительного и аудиального восприятия с работой пишущей руки.
Ребенок с такими проблемами не ползает, тяжело начинает ходить, с большим трудом начинает читать и писать, воспринимая информацию на слух или зрительно. У детей с данной патологией, если вовремя не начать коррекцию и последующую реабилитацию, возникает целый ряд серьезных проблем, которые являются серьезным препятствием в развитии и обучении, в том числе и школьном.
В том случае, если агенезия мозолистого тела не сопровождается никакими другими патологиями развития, прогноз для больного достаточно благоприятный.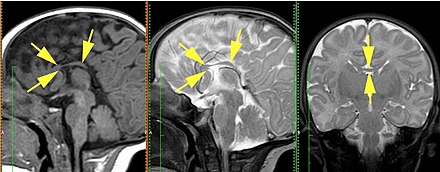
Агенезия мозолистого тела, хоть и встречается сравнительно часто, тем не менее, является малоизученным состоянием, особенно на просторах нашей страны.
Перелыгин Демьян — Благотворительный фонд Алёша!
До сбора
В семье Перелыгиных мечтали о сыне и к запланированной беременности отнеслись со всей серьезностью.
Увы, чуда, которого так ждали родители, не произошло. После родов подозрения врачей подтвердились, а к уже поставленным диагнозам добавилось еще немало сопутствующих: глухота, атрофия зрительного нерва, птоз верхнего века, водянка обоих яичек, синдром нарушения мышечного тонуса, ВПР, ВПС. Мальчика перевели в отделение патологии новорожденных, обследовали, сделали КТ и рекомендовали маме искать невролога и начинать лечение.
По рекомендации невролога Деме сделали МРТ и исследование когнитивных вызванных потенциалов головного мозга. Результаты выявили медленную проводимость информации, грубую психоречевую задержку, но также показали, что агенезия мозолистого тела у Демьяна лишь частичная. Невролог назначил курс Томатиса и микрополяризацию, а также препараты для стимуляции головного мозга. Деме нужны постоянные реабилитации, но родителям катастрофически не хватает средств на их оплату.
Невролог назначил курс Томатиса и микрополяризацию, а также препараты для стимуляции головного мозга. Деме нужны постоянные реабилитации, но родителям катастрофически не хватает средств на их оплату.
Благодаря вашим пожертвованиям всего за месяц необходимая сумма была собрана, и теперь Демьян сможет продолжить свой путь к восстановлению и обретению новых навыков. Спасибо всем!
Обращение семьи
MEDISON.RU — Нейросонография в диагностике церебральный поражений у детей с врожденной вирусной инфекцией
УЗИ сканер RS80

Введение

Цель настоящего исследования — выявление особенностей структурных изменений головного мозга у новорожденных при различных внутриутробных вирусных инфекциях.
Материалы и методы
Под динамическим наблюдением находились 50 детей с врожденной вирусной инфекцией, госпитализированных в отделения патологии, реанимации и интенсивной терапии городской клинической больницы N4 Ижевска. Этиологический диагноз был установлен на основании клинических, иммунологических (иммуноферментный анализ) исследований, реакции непрямой и прямой иммунофлюоресценции. При вирусологическом исследовании иммунофлюоресцентным методом определен смешанный характер врожденной вирусной инфекции в 100% случаев (вирусно-вирусные ассоциации состояли из двух и более антигенов). Антигены энтеровирусов (Коксаки А и В, энтеро 68-71, полиомиелит) обнаружены в 100% случаев, цитомегалии — в 86%, краснухи — в 52,1%, герпеса простого — в 40%.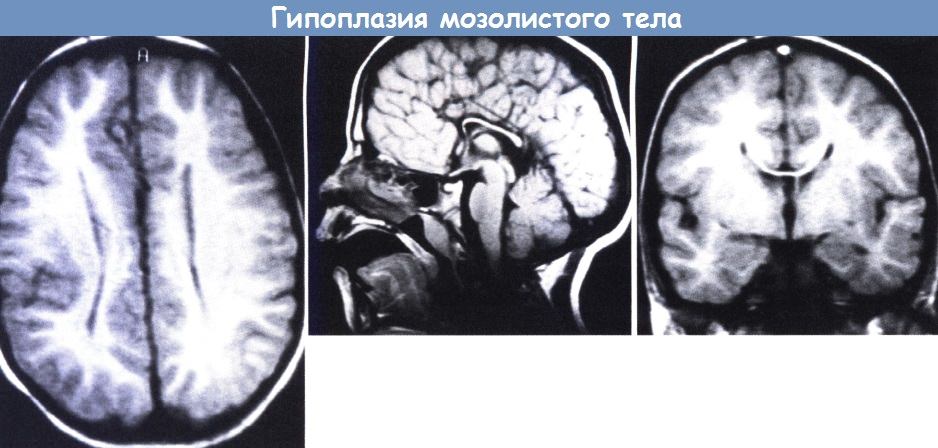
Эхографическое обследование новорожденных проводили при помощи современных ультразвуковых приборов. Для нейросонографии использовали конвексный датчик 5МГц. Сканирование осуществляли в стандартных плоскостях: коронарной, сагиттальной, парасаггитальной и аксиальной. Анализ эхограммы включал в себя оценку состояния паренхимы головного мозга, желудочковой системы, цистерн и субарахноидального пространства, рисунка извилин и борозд, пульсации мозговых сосудов и наличие очаговых патологических образований. Эхографическое исследование проводили наблюдаемым детям на первой неделе жизни и в динамике.
Всего выполнено 100 эхографических исследований головного мозга, при которых патологические изменения диагностированы у 36 (72%) детей. Внутричерепные кровоизлияния обнаружены у 27 (84,37%) детей : перивентрикулярные кровоизлияния — у 19, внутрижелудочковые — у 8.
В 2 случаях перивентрикулярное кровоизлияние сочеталось с внутрижелудочковым. Перивентрикулярная лейкомаляция выявлена у 3 (6,3%) больных. Врожденные пороки развития ЦНС при ультразвуковом исследовании диагностированы у 7 (21,9%) новорожденных: врожденная гидроцефалия — у 5, частичная агенезия мозолистого тела — у 1, голопрозэнцефалия (семилобарная форма) — у 1.
Перивентрикулярная лейкомаляция выявлена у 3 (6,3%) больных. Врожденные пороки развития ЦНС при ультразвуковом исследовании диагностированы у 7 (21,9%) новорожденных: врожденная гидроцефалия — у 5, частичная агенезия мозолистого тела — у 1, голопрозэнцефалия (семилобарная форма) — у 1.
На эхограмме ребенка с частичной агенезией мозолистого тела визуализировались узкие, расположенные далеко друг от друга, передние рога боковых желудочков с нечетко контурированными стенками и вогнутым наружным краем. III желудочек был расширен до 5 мм. Полость прозрачной перегородки и межпоясная борозда не визуализировались. Наблюдалось также веерообразное, радиальное расположение борозд (рис. 1).
Рис. 1. Частичная агенезия мозолистого тела:
1 — III желудочек, 2 — веерообразное расположение извилин.
У 9 детей диагностированы ПВK I степени; субэпендимальная гематома визуализировалась в виде гиперэхогенной структуры овальной формы в проекции хвостатого ядра и каудоталамической вырезки, размерами от 2 до 7 мм в диаметре. Перивентрикулярные кровоизлияния II степени выявлены у 5 детей и были представлены гиперэхогенными участками (тромбами) более 1 см в диаметре. Перивентрикулярные кровоизлияния Ш степени (рис. 2) обнаружены нами у 4 детей, они были двусторонними, с наличием тромбов внутри боковых желудочков.
Перивентрикулярные кровоизлияния II степени выявлены у 5 детей и были представлены гиперэхогенными участками (тромбами) более 1 см в диаметре. Перивентрикулярные кровоизлияния Ш степени (рис. 2) обнаружены нами у 4 детей, они были двусторонними, с наличием тромбов внутри боковых желудочков.
Рис. 2. Перивентрикулярное кровоизлияние III степени. Постгеморрагическая внутренняя гидроцефалия вследствие расширения всех отделов желудочковой системы.
Перивентрикулярные кровоизлияния IV степени выявлено на нейросонографии только у одного из наблюдаемых детей. На эхограмме визуализировалось гиперэхогенное образование с четкими контурами, расположенное над телом бокового желудочка (паренхиматозное кровоизлияние), в динамике отмечалась неоднородная эхоструктура даннного образования с формированием в дальнейшем порэнцефалической псевдокисты. При повторных ультразвуковых исследованиях у 14 (38,9%) детей субэпендимальная гематома уменьшилась в размерах, отмечалась ее неоднородность, у 2 детей субэпендимальная гематома разрешилась. У 3 детей на месте субэпендимальной гематомы образовалась анэхогенная структура округлой формы с четкими ровными контурами (псевдокиста).
При повторных ультразвуковых исследованиях у 14 (38,9%) детей субэпендимальная гематома уменьшилась в размерах, отмечалась ее неоднородность, у 2 детей субэпендимальная гематома разрешилась. У 3 детей на месте субэпендимальной гематомы образовалась анэхогенная структура округлой формы с четкими ровными контурами (псевдокиста).
У 7 новорожденных выявлены одна или несколько (2-4) изолированных кистозных структур диаметром от 2 до 10 мм, локализующихся в теле и в верхушке сосудистых сплетений боковых желудочков. Эти патологические изменения были определены в одном боковом желудочке у 5 детей, в обоих боковых желудочках — у 2. Исходные данные вентрикулометрии соответствовали нормальным значениям у 5 (13,9%) пациентов. В 2 случаях обнаружено расширение межполушарной щели и субарахноидальных пространств по конвекситальной поверхности мозга, а также умеренное симметричное увеличение ширины лобных рогов и высоты тел боковых желудочков (до 7 мм). В конце периода наблюдения у 4 (11,1%) больных кисты сосудистых сплетений имели прежние размеры, а данные вентрикулометрии соответствовали возрастной норме. При динамическом наблюдении у 2 детей было отмечено прогрессирующее расширение боковых желудочков (с 7 до 15 мм). Дальнейшее расширение межполушарной щели и субарахноидальных пространств по конвекситальной поверхности мозга от 5 до 8 мм было выявлено в 4 случаях. У 1 пациента кисты сосудистых сплетений при повторной эхоэнцефалографии не обнаружены.
При динамическом наблюдении у 2 детей было отмечено прогрессирующее расширение боковых желудочков (с 7 до 15 мм). Дальнейшее расширение межполушарной щели и субарахноидальных пространств по конвекситальной поверхности мозга от 5 до 8 мм было выявлено в 4 случаях. У 1 пациента кисты сосудистых сплетений при повторной эхоэнцефалографии не обнаружены.
У всех наблюдаемых нами детей с перивентрикулярными кровоизлияниями также обнаружены субэпендимально расположенные кистозные структуры, локализующиеся на уровне каудоталамической вырезки, диаметром от 3 до 8 мм. У большинства новорожденных (88,2%) субэпендимально расположенные кисты являлись единой полостью с однородным или неоднородным содержимым и в 4 случаях — состояли из множества жидкостных включений, окруженных эхопозитивным ободком («соты»). У 10 детей субэпендимальные кисты располагались в одном полушарии, у 7 детей — в обоих полушариях. Помимо указанных выше субэпендимальных кист, у 3 больных обнаружены перегородочные структуры в передних рогах боковых желудочков и неравномерное распределение ликвора.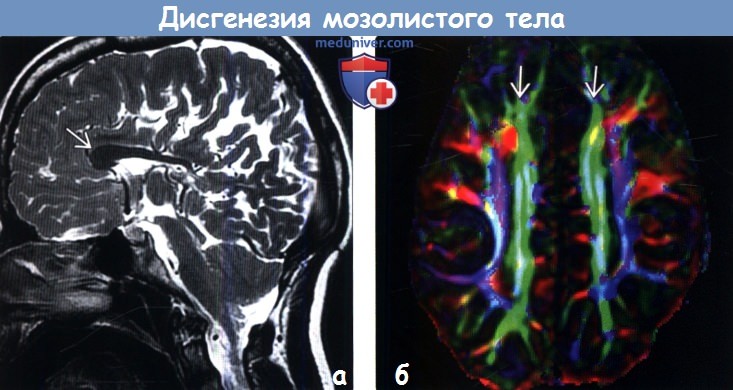 Во всех перечисленных случаях исходные данные вентрикулометрии соответствовали нормальным значениям. Однако при динамической эхоэнцефалографии у 7 (19,4%) детей отмечалось умеренное (до 9 мм), симметричное увеличение ширины лобных рогов и высоты тел боковых желудочков; асимметричная умеренная дилатация боковых желудочков — у 2 пациентов. При дальнейшем наблюдении субэпендимальные кисты уменьшились в размерах у большинства детей (70,6 %) и резорбировались — в 5 случаях. Внутрижелудочковые кровоизлияния диагностированы в 8 (22,2%) случаях : у 6 недоношенных и у 2 доношенных детей. Преобладали односторонние ВЖК I степени (65%), двусторонние были определены в 35% случаев. Постгеморрагическая дилатация боковых желудочков обнаружена у всех детей. Вентрикуломегалия тяжелой степени постгеморрагического генеза выявлена только у 1 ребенка и сопровождалась увеличением глубины тел более 20 мм и расширением всех отделов боковых желудочков (рис. 3).
Во всех перечисленных случаях исходные данные вентрикулометрии соответствовали нормальным значениям. Однако при динамической эхоэнцефалографии у 7 (19,4%) детей отмечалось умеренное (до 9 мм), симметричное увеличение ширины лобных рогов и высоты тел боковых желудочков; асимметричная умеренная дилатация боковых желудочков — у 2 пациентов. При дальнейшем наблюдении субэпендимальные кисты уменьшились в размерах у большинства детей (70,6 %) и резорбировались — в 5 случаях. Внутрижелудочковые кровоизлияния диагностированы в 8 (22,2%) случаях : у 6 недоношенных и у 2 доношенных детей. Преобладали односторонние ВЖК I степени (65%), двусторонние были определены в 35% случаев. Постгеморрагическая дилатация боковых желудочков обнаружена у всех детей. Вентрикуломегалия тяжелой степени постгеморрагического генеза выявлена только у 1 ребенка и сопровождалась увеличением глубины тел более 20 мм и расширением всех отделов боковых желудочков (рис. 3).
Рис.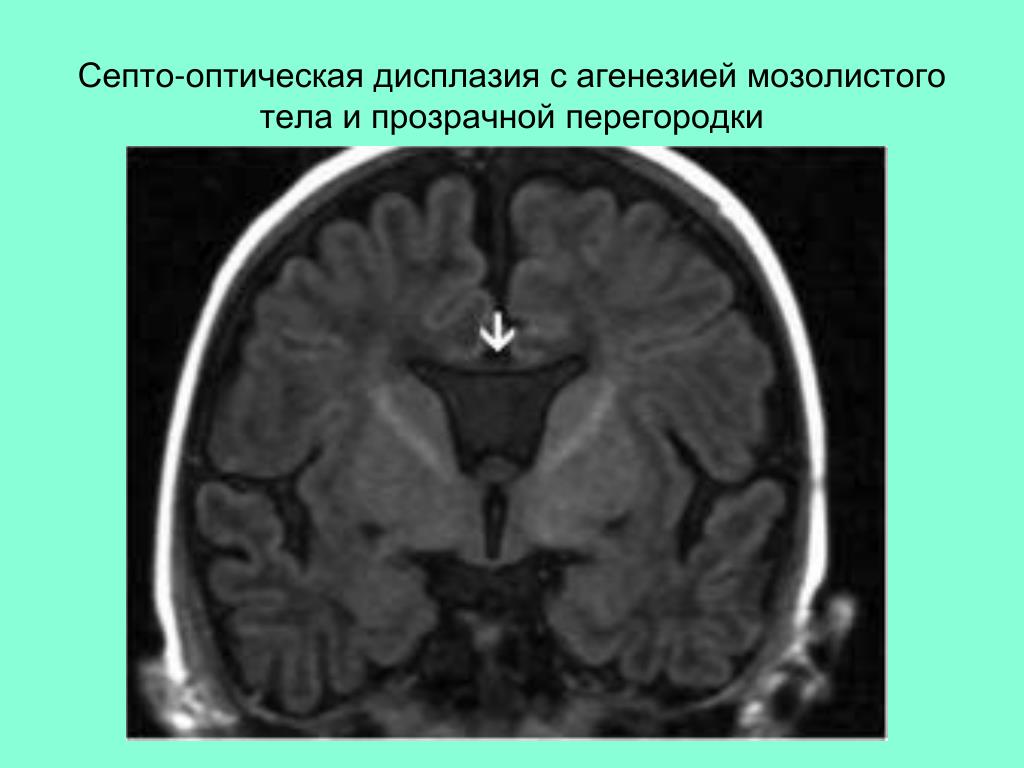 3. Вентрикуломегалия тяжелой степени, вентрикулит:
3. Вентрикуломегалия тяжелой степени, вентрикулит:
1 — гиперэхогенная взвесь в полостях расширенных боковых желудочков, 2 — фибриновые тяжи в расширенных боковых желудочках, 3 — уплотнение стенки боковых желудочков (эпендиматит).
У 5 (10%) детей диагностированы вирусные менингиты и менингоэнцефалиты, осложнившиеся в дальнейшем развитием постинфекционной вентрикуломегалии и расширением субарахноидального пространства. При динамическом наблюдении у 1 ребенка на нейросонографии и компьютерной томографии была выявлена внутренняя гидроцефалия с атрофией мозга. Вентрикулит диагностирован у 1 ребенка с менингоэнцефалитом, на эхограмме отмечалось утолщение стенок боковых желудочков, наличие в расширенных боковых желудочках гиперэхогенной взвеси и расширение сосудистых сплетений. При повторном сканировании после санации ликвора наблюдалось уменьшение вентрикуломегалии и исчезновение гиперэхогенных включений.
Перивентрикулярная лейкомаляция обнаружена у 3 новорожденных и сопровождалась зоной повышенной эхогенности, окружающей оба желудочка вокруг тел и затылочных рогов. При динамическом наблюдении у 1 ребенка — кистозная дегенерация мозга в области повышенной эхогенности с образованием множественных перивентрикулярных псевдокист размерами от 2 до 5 мм в диаметре (рис. 4 а,б). У всех детей с перивентрикулярной лейкомаляцией в дальнейшем имела место умеренная симметричная вентрикуломегалия и расширение субарахноидального пространства.
Рис. 4. Перивентрикулярная лейкомаляция.
а) Стадия образования псевдокист (стрелки) в коронарной плоскости.
б) Стадия образования псевдокист (стрелки) в сагиттальной плоскости.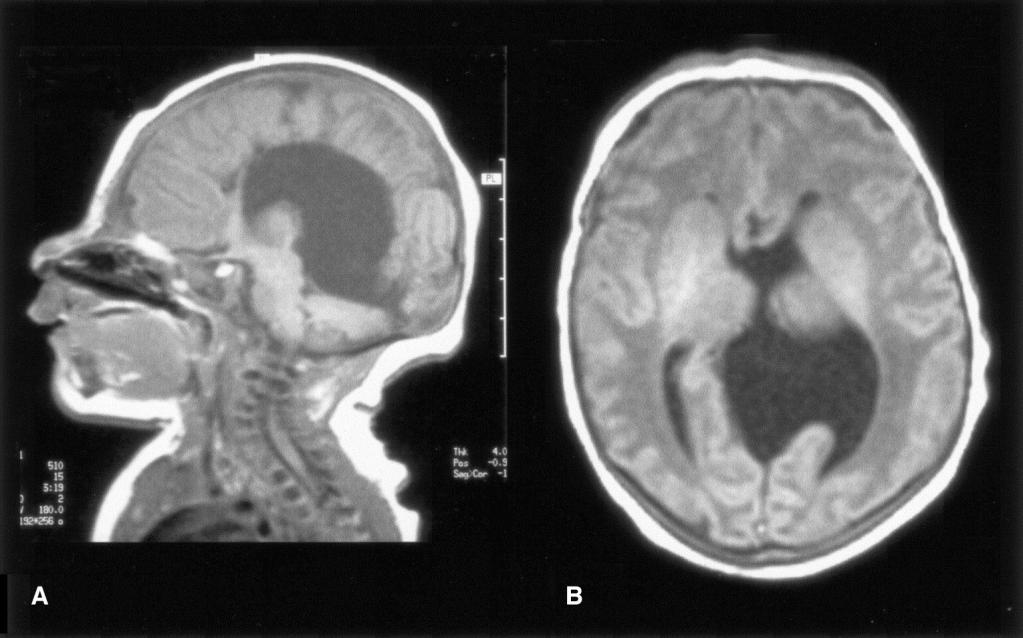
У 25 (69,4 %) новорожденных детей были выявлены эхопризнаки незрелости мозга: слабая выраженность извилин, визуализация полости Верге и полости прозрачной перегородки в виде срединно расположенных анэхогенных структур, повышенная эхогенность сильвиевой борозды и визуализация ее в виде «треугольника».
Следует отметить, что нами не была установлена отчетливая связь между числом, локализацией, динамикой субэпендимальных кист и данными комплексного вирусологического и иммунологического обследования, а также клиническими проявлениями как в раннем неонатальном периоде, так и в конце периода наблюдения. Диагностированные эхографические изменения у наблюдаемых детей не являлись специфичными и были полиморфны. Структурные изменения головного мозга, такие как внутричерепные кровоизлияния и перивентрикулярная лейкомаляция встречались при всех вышеперечисленных вирусных моноинфекциях. Врожденные пороки развития, менингит и менингоэнцефалит диагностированы у детей со смешанной вирусной инфекцией (энтеровирусной в ассоциации с краснушной, герпетической и цитомегаловирусной инфекцией). Полиморфность и неспецифичность структурных изменений головного мозга могут быть объяснены смешанным характером вирусной инфекции.
Врожденные пороки развития, менингит и менингоэнцефалит диагностированы у детей со смешанной вирусной инфекцией (энтеровирусной в ассоциации с краснушной, герпетической и цитомегаловирусной инфекцией). Полиморфность и неспецифичность структурных изменений головного мозга могут быть объяснены смешанным характером вирусной инфекции.
Литература
- Ватолин К.В. Ультразвуковая диагностика заболеваний головного мозга у детей. — М.: Видар. — 1995. — 129 с.
- Геппе Н.А., Нестеренко О.С., Нагибина Н.С. и др. Пороки развития ЦНС у новорожденных с внутриутробной инфекцией // Педиатрия. — 1999. — N5. — С. 42-44.
- Доманин Е.И., Волосников Д.К., Масленникова Н.В. и др. Частота пороков головного мозга у новорожденных // Росс. вестник перинатологии и педиатрии. — 2000. — т.45, N2. — С. 28-31.
- Зубарева Е.А., Неижко Л.Ю. Клиническая нейросонография новорожденных детей и детей раннего возраста // Клиническое руководство.
 Под ред. В.В. Митькова, М.В. Медведева. — М.: Видар. — 1997. — Т.3. — С.9-72.
Под ред. В.В. Митькова, М.В. Медведева. — М.: Видар. — 1997. — Т.3. — С.9-72. - Озерова О.Е., Кудашов Н.И., Орловская И.В. и др. УЗ-особенности структурных изменений головного мозга новорожденных с внутриутробной герпес-цитомегаловирусной инфекцией // SonoAce International. — 2000. — вып. 6. — С.44-49.
- Reuck J., Charrha A., Richardson E. Pathogenesis and evolution of periventricular leukomalacia in infancy // Arch. Neurol. — 1972, Vol. 27. — N9. — P. 229-236.
- Zorzi C., Angonese I. Subependimal pseudocysts in the neonate // Eur. J. Pediatr. — 1989. — Vol.148. — P.462-464.
 Под ред. В.В. Митькова, М.В. Медведева. — М.: Видар. — 1997. — Т.3. — С.9-72.
Под ред. В.В. Митькова, М.В. Медведева. — М.: Видар. — 1997. — Т.3. — С.9-72.УЗИ сканер RS80
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
что это такое, причины, последствия
Агенезия (недоразвитие) мозолистого тела (АМТ) – одна из форм церебральных дисгенезий (нарушение развития), обусловленных нарушением формирования плода во внутриутробный период. Выделяют тотальную и парциальную формы. В первом случае отсутствуют комиссуральные (нервные пучки, соединяющие гемисферы) волокна. Во втором случае недоразвиты каудальные (задние) и ростральные (передние) сегменты мозолистого тела. АМТ часто (около 70% случаев) встречается в сочетании с другими пороками развития (чаще патологии сердечно-сосудистой, мочеполовой и опорно-двигательной системы) или изолированно.
Характеристика
Агенезия – это такая патология, которая характеризуется утратой органом способности к развитию, что приводит к уменьшению размеров и нарушению функций. Мозолистое тело – это мозговой отдел, который соединяет между собой полушария, его агенезия сопровождается нарушением высших функций психики. Мозолистое тело – сплетение волокон нервной ткани. Количество волокон превышает 250-300 миллионов.
Мозолистое тело – сплетение волокон нервной ткани. Количество волокон превышает 250-300 миллионов.
В норме мозговой отдел представлен в форме полосы шириной около 8 см, состоящей из отростков аксонов, располагается под корковым слоем. Его волокна проходят поперечно, соединяя участки гемисфер, находящиеся симметрично. Некоторые волокна соединяют участки, расположенные в разных зонах разных гемисфер, например, лобные зоны правого полушария с теменными или затылочными зонами левого.
Диагноз агенезия мозолистого отдела означает, что между гемисферами частично или полностью отсутствуют соединяющие нервные волокна. Распространенность заболевания составляет около 0,7% в общей популяции, около 3% среди больных с диагностированной умственной отсталостью. Наличие межполушарных связей обеспечивает взаимодействие мозговых структур обеих гемисфер.
При сегментарном или полном отсутствии мозолистого тела у новорожденного взаимодействие нарушается. Как показывают научные эксперименты, через комиссуры мозолистого тела передаются частично обработанные в корковом слое сенсорные данные..jpg) Мозолистое тело меньше участвует в переносе команд, управляющих двигательной активностью, больше – в межполушарной передаче вербальной информации и зрительно-пространственных данных.
Мозолистое тело меньше участвует в переносе команд, управляющих двигательной активностью, больше – в межполушарной передаче вербальной информации и зрительно-пространственных данных.
С увеличением сложности выполняемых интеллектуальных задач повышается активность структур мозолистого тела. Важная функция тела – торможение мозговых процессов для лучшей дифференциации работы отделов разных гемисфер, что обеспечивает более эффективную обработку информации. Исследования показывают, качество взаимодействия между структурами разных гемисфер влияет на уровень интеллекта.
Аплазия (отсутствие части или всего органа), истончение или утолщение отдела мозолистого тела – варианты аномального формирования. Если отдел мозга отсутствует или нарушается процесс его нормального развития, это может стать причиной возникновения таких патологий, как шизофрения, аутизм, дислексия (нарушение способности приобретать навыки чтения и письма при сохранении способности к обучению), синдром, проявляющийся недостатком внимания и гиперактивностью.
Например, при рано дебютирующих формах шизофрении в ходе инструментального обследования выявляется истончение структур тела. При поздно дебютирующих формах шизофрении с относительно благоприятным прогнозом течения выявляется утолщение его структур.
Прямая связь аномалий формирования этого отдела мозга с развитием психозов не установлена. Однако считается, что частичная агенезия (недоразвитие) мозолистого тела может играть роль пускового механизма в развитии психотических расстройств. В числе сочетанных пороков формирования структур ЦНС часто выявляются кистозные образования (арахноидальные, межполушарные кисты), синдромы Арнольда-Киари, Денди-Уокера, аномальное развитие извилин. В 80% случаев АМТ сочетается с гидроцефалией.
Причины возникновения
Агенезия (нарушение формирования или отсутствие) мозолистого тела головного мозга у ребенка – это врожденная аномалия, которая с частотой около 30% встречается среди пациентов с генетически обусловленными, наследственными синдромами. В числе причин возникновения патологии стоит отметить митохондриальные заболевания, мутации генов, цитогенетические синдромы.
В числе причин возникновения патологии стоит отметить митохондриальные заболевания, мутации генов, цитогенетические синдромы.
АМТ встречается в клинической картине болезни Гентингтона (нейродегенеративное заболевание хронического течения, характеризующееся прогрессирующей гибелью нервных клеток) и метаболических расстройств наследственной формы. В большинстве случаев наследственные синдромы с участием в патогенезе АМТ – мультисистемные (происходит поражение многих органов и систем). Провоцирующие факторы:
- Заболевания, перенесенные матерью в период гестации (инфекции, передающиеся половым путем, респираторные вирусные болезни).
- Интоксикации (хронические, острые).
- Неблагоприятное течение беременности (токсикоз в I, II триместре гестации, угроза прерывания).
В структуре причин врожденных пороков формирования мозолистого отдела и других органов выделяют случайные повреждающие факторы, на которые приходится 60% случаев аномалий развития. В 10% случаев причиной возникновения аномалий становятся хромосомные болезни, в 20% случаев – тератогенное (нарушающее эмбриональный морфогенез) воздействие фармацевтических препаратов. Внутриутробные инфекции (преимущественно вирусные) приводят к возникновению врожденных пороков в 10% случаев.
Внутриутробные инфекции (преимущественно вирусные) приводят к возникновению врожденных пороков в 10% случаев.
Клинические проявления
Изолированная форма агенезии (недоразвитие) мозолистого тела – это такая патология, которая может протекать бессимптомно, что затрудняет раннюю диагностику. Неврологический дефицит чаще обусловлен сопутствующими патологиями. При наличии сочетанных патологий у 40% пациентов выявляются двигательные расстройства.
У 30% больных детей наблюдается задержка умственного, речевого и физического развития. Умственная отсталость диагностируется у 70% пациентов с нарушением формирования мозолистого отдела. Ведущий признак у младенцев 1-го года жизни – судорожный синдром. Другие симптомы агенезии (недоразвития) мозолистого тела, характерные для новорожденных до 1 года:
- Нарушение восприятия и сенсорных реакций.
- Ухудшение коммуникабельности, отсутствие психологической связи с матерью.
- Недостаточная модуляция (изменение силы, тональности и других характеристик) крика.
У детей старшего возраста наблюдаются признаки: расстройство терморегуляции (гипотермия – понижение показателей температуры тела ниже отметки 35°C, гипертермия – устойчивое повышение показателей температуры тела), нарушение двигательной координации и пространственной ориентации, ухудшение памяти (зрительной, слуховой). Например, в ходе исследований у детей 4-8 лет обнаруживается нарушение тактильного восприятия – ребенок затрудняется назвать предмет, который ощупывает субдоминантной (не ведущей) рукой.
Пациенты испытывают затруднения при тактильном (на ощупь) прохождении лабиринта, что связано с нарушением передачи пространственной информации межмануального (между конечностями) характера. У детей с диагностированной патологией мозолистого тела отмечается ухудшение социальной адаптации и навыков планирования.
Одновременно наблюдаются симптомы: бедность эмоций, эмоциональная незрелость (неспособность адекватно реагировать на бытовые проблемы и жизненные ситуации, неумение действовать с учетом целесообразности). Став взрослыми, такие люди часто страдают от одиночества.
Став взрослыми, такие люди часто страдают от одиночества.
Диагностика
Недоразвитие мозолистого тела выявляется в ходе инструментального обследования головного мозга, которое включает такие методы, как МРТ, КТ, нейросонография. Диагностика в формате МРТ, КТ часто показывает наличие кистозного образования в межполушарной зоне, увеличение диаметра III желудочка, патологическое изменение геометрической формы боковых желудочков.
Агенезия (недоразвитие или отсутствие) мозолистого тела у плода выявляется в ходе пренатального ультразвукового обследования. При наличии сочетанных врожденных пороков развития и хромосомных дефектов патология может стать поводом для прерывания беременности. Период проведения пренатального скрининга – II, III триместр гестации.
Лечение
Специфический протокол лечения АМТ не разработан. Коррекция судорожных, эпилептических приступов осуществляется согласно общим принципам при помощи противосудорожных, противоэпилептических препаратов.
Последствия
Если в ходе пренатальной диагностики выявляется сочетание АМТ с хромосомными дефектами и аномалиями развития органов и систем, прогноз неблагоприятный. Если речь идет об изолированной форме АМТ, считается, что ситуация не требует коррекции акушерской тактики. Последствия агенезии (недоразвития) мозолистого тела проявляются в нарушении функций головного мозга.
Возможные последствия: апраксия (нарушение целенаправленных движений, нарушение деятельности, связанной с необходимостью планирования и следования плану), судорожные приступы, ухудшение психической деятельности, гипертермия (перегревание тела), гипотермия (переохлаждение тела), ухудшение способности ориентироваться в пространстве. Прогноз составляется индивидуально, зависит от наличия, вида, характера течения сопутствующих патологий.
Агенезия (недоразвитие, отсутствие) мозолистого тела – врожденный порок формирования мозговых структур. Изолированная форма сопровождается нарушениями функций мозга. При сочетанной форме прогноз неблагоприятный, возможные последствия – инвалидность, летальный исход.
Просмотров: 230
УЗИ. Нормальное развитие головного мозга и пороки развития мозолистого тела. Лекция для врачей
Лекция для врачей «УЗИ. Нормальное развитие головного мозга и пороки развития мозолистого тела». Лекцию для врачей проводит Михаил Борисов Lis Maternity Hospital (Израиль)
Дополнительный материал
ПОРОКИ РАЗВИТИЯ ГОЛОВНОГО МОЗГА
Пороки развития могут проявляться в различных сочетаниях. Так, например, при синдроме Дуранда-Дзунина признаки дизрафии сочетаются с гидроцефалией, сопровождающейся увеличением мозгового черепа, агенезией прозрачной перегородки, расщеплением дужек позвонков, искривлением стоп и двусторонней гипоплазией почек, ведущей к нарушению водного обмена. Синдром имеет семейный, по-видимому, наследственный характер. Описали его в 1955 г. итальянские педиатры S. Durand и F. Zunin.
Синдром имеет семейный, по-видимому, наследственный характер. Описали его в 1955 г. итальянские педиатры S. Durand и F. Zunin.
В особую группу аномалий развития могут быть выделены выраженные вторичные врожденные пороки развития черепа и мозга, возникшие в различные периоды онтогенеза. Причины таких аномалий многообразны: заболевания матери в период беременности, облучение, травматические повреждения плода, воздействие на плод разнообразных токсических факторов, в частности алкоголя и многочисленных лекарственных препаратов, оказывающих тератогенное действие. Пороки развития ЦНС являются следствием одного или нескольких основных патологических процессов, нарушающих развитие мозга: образование нервной трубки, разделение ее краниального отдела на парные образования, миграция и дифференциация клеточных элементов нервной ткани. Они могут проявляться на трех уровнях: клеточном, тканевом и органном.
Ниже приводится описание некоторых дефектов развития головного мозга и черепа, возникающих в процессе онтогенеза (вследствие дизэмбриогенеза).
Анэнцефалия — отсутствие большого мозга, костей свода черепа и покрывающих его мягких тканей. На месте мозгового вещества обычно располагается соединительная ткань, богатая кровеносными сосудами, с кистозными полостями, выстланными медуллярным эпителием, глиальная ткань, единичные нервные клетки, остатки сосудистых сплетений.
Эксэнцефалия — отсутствие костей свода черепа (акрания) и мягких покровов головы, в результате чего большие полушария располагаются открыто на основании черепа в виде отдельных узлов, покрытых мягкой мозговой оболочкой.
Гидроанэнцефалия — полное или почти полное отсутствие больших полушарий при сохранности костей свода черепа и его покровных тканей. Голова при этом нормальных размеров или несколько увеличена. Полость черепа заполнена главным образом ЦСЖ. Продолговатый мозг и мозжечок достаточно развиты. Средний мозг и другие отделы головного мозга могут отсутствовать или представлены рудиментарно. Впервые эта форма порока была описана Ж. Крювелье в 1835 г. под названием «гидроцефалическая анэнцефалия».
Порэнцефалия истинная — наличие в ткани конечного мозга полостей разных размеров, выстланных эпендимой и сообщающихся с желудочковой системой и субарахноидальным пространством.
Порэнцефалия ложная — замкнутые полости в большом мозге, не имеющие эпендимной выстилки и представляющие собой кисты после энцефаломаляции разного происхождения.
Кистозная дисплазия головного мозга, или полипорэнцефалия, — врожденная дисплазия больших полушарий головного мозга, характеризующаяся образованием в нем множественных полостей, обычно сообщающихся с желудочковой системой мозга.
Прозэнцефалия — порок развития, при котором большие полушария мозга отделяет друг от друга лишь мелкая продольная борозда, поэтому граница между правой и левой половинками конечного мозга нечеткая (встречается с частотой 1:16 000).
Голопрозэнцефалия — порок развития мозга, при котором его большие полушария не разделены и имеют вид единой полусферы, а боковые желудочки представлены единой полостью. Часто сочетается с другими врожденными пороками. Обычно смерть наступает вскоре после рождения. Может быть проявлением трисомии хромосом 13-15. Пороки конечного мозга сопровождаются различными, порой грубыми, нарушениями строения лица и его костей, в частности цебоцефалией, этмоцефалией и циклопией. Дети с циклопией обычно рождаются мертвыми.
Агирия (лиссэнцефалия) — недоразвитие извилин больших полушарий, при этом поверхность их сглажена (гладкий мозг). При микроскопии выявляются грубое изменение архитектоники коры больших полушарий, отсутствие в ней обычных клеточных слоев. Проявляется выраженным нарушением психомоторного развития, полиморфными судорогами, парезами или параличами. Дети обычно умирают в течение первого года жизни.
Микро- и полигирия — порок, при котором на поверхности больших полушарий имеется множество беспорядочно расположенных мелких извилин.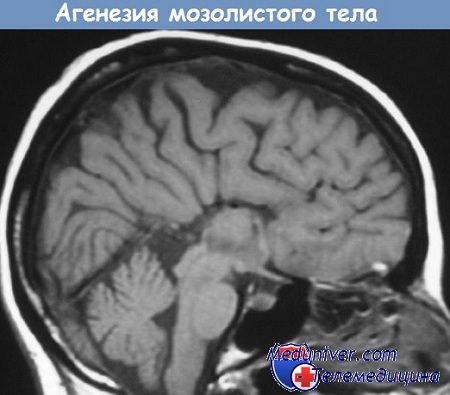 Обычно микрогирия проявляется симметрично и сопровождается нарушением послойного строения коры, имеющей не более 4 слоев.
Обычно микрогирия проявляется симметрично и сопровождается нарушением послойного строения коры, имеющей не более 4 слоев.
Пахигирия (макрогирия) — укрупнение основных извилин, тогда как вторичные и третичные извилины отсутствуют, борозды при этом выпрямлены, они короткие и неглубокие. Цитоархитектоника коры в таких случаях нарушена. В белом веществе мозга встречаются гетеротопии нервных клеток.
Гипоплазия, или аплазия (агенезия), мозолистого тела — частичное или полное отсутствие мозолистого тела. В случае его аплазии III желудочек мозга остается открытым. Если отсутствует лишь задняя спайка, а само мозолистое тело только укорочено, то это называется гипоплазией.
Синдром Айкарди — гипоплазия мозолистого тела в сочетании с другими пороками, в частности с хориоретинальными аномалиями, при этом характерны спазмы сгибательной мускулатуры или миоклонические приступы, множественные лакунарные очаги в сосудистой и сетчатой оболочках глаз, выявляемые при офтальмоскопии в перипапиллярной зоне. Размеры атрофических хориоретинальных очагов варьируют от небольших, меньше поперечника диска зри- тельного нерва, до диаметра в несколько его поперечников. Часто имеются дизрафические изменения позвоночника. Возможны умственная отсталость, маятникообразный нистагм, аномалии развития глаз (микрофтальм, колобомы зрительного нерва и хориоидальной оболочки, эктазия склеры и др.). Описан синдром только у девочек, это позволяет полагать, что болезнь может быть следствием мутации в Х-хромосоме, которая является летальной при развитии мужского организма. Описал в 1956 г. французский педиатр J. Aicardi.
Размеры атрофических хориоретинальных очагов варьируют от небольших, меньше поперечника диска зри- тельного нерва, до диаметра в несколько его поперечников. Часто имеются дизрафические изменения позвоночника. Возможны умственная отсталость, маятникообразный нистагм, аномалии развития глаз (микрофтальм, колобомы зрительного нерва и хориоидальной оболочки, эктазия склеры и др.). Описан синдром только у девочек, это позволяет полагать, что болезнь может быть следствием мутации в Х-хромосоме, которая является летальной при развитии мужского организма. Описал в 1956 г. французский педиатр J. Aicardi.
Микроцефалия (синдром Джакомини) — недоразвитие головного мозга, проявляющееся при рождении уменьшением его массы и размеров (рис. 24.7). Микроцефалия обычно сочетается с уменьшенной окружностью головы (не менее чем на 5 см от средних показателей) и дальнейшим отставанием роста мозгового черепа (микрокрания), при этом швы его могут длительно оставаться открытыми. Кости черепа часто утолщены, в них рано формируются диплоидные каналы, внутричерепное давление не повышено. При микрокрании обычно отмечается соответствующее уменьшение размеров и массы головного мозга — микроцефалия. Морфологическим ее признаком является недоразвитие и неправильное строение больших полушарий при сравнительно нормальной архитектонике мозжечка и ствола мозга. Ребенок с микроцефалией обычно отстает в умственном, а зачастую и в физическом развитии.
При микрокрании обычно отмечается соответствующее уменьшение размеров и массы головного мозга — микроцефалия. Морфологическим ее признаком является недоразвитие и неправильное строение больших полушарий при сравнительно нормальной архитектонике мозжечка и ствола мозга. Ребенок с микроцефалией обычно отстает в умственном, а зачастую и в физическом развитии.
Микроцефалия может быть первичной (истинной, генетически обусловленной) и вторичной. Первичная микроцефалия — следствие генетического дефекта, наследуемого по аутосомно-рецессивному типу или возникающего в связи с хромосомными аномалиями. Вторичная микроцефалия может быть обусловлена перенесенной внутриутробно инфекцией (краснуха, цитомегаловирусный энцефалит, токсоплазмоз), интоксикаций или асфиксией, травмой мозга. При вторичной микроцефалии в мозге возможны кистозные полости, очаги кровоизлияния и обызвествления. Внешний вид детей с микроцефалией своеобразен и характеризуется диспропорцией между размерами мозгового черепа и лица. Частота микроцефалии среди новорожденных 1:5000. Среди всех случаев олигофрении 11% отмечается у больных с микроцефалией.
Частота микроцефалии среди новорожденных 1:5000. Среди всех случаев олигофрении 11% отмечается у больных с микроцефалией.
Рис. 24.7. Микроцефалия у ребенка 3 лет.
Макроцефалия — увеличение массы и объема головного мозга, а вместе с этим и мозгового черепа при рождении, встречается значительно реже микроцефалии. В большинстве случаев сопровождается нарушением расположения мозговых извилин, изменениями цитоархитектоники коры, очагами гетеротопии в белом веществе, при этом обычно отмечаются проявления олигофрении, возможен судорожный синдром. Причиной макроцефалии может быть поражение паренхимы мозга (липоидозы). На краниограммах костные швы не расширены, желудочки мозга нормального или почти нормального размера. Макроцефалию следует дифференцировать от гидроцефалии.
Возможна частичная макроцефалия (увеличение одного из больших полушарий), которая обычно сочетается с асимметрией мозгового черепа. Гемигипертрофия черепа за счет выбухания с одной стороны чешуи височной кости и прилежащих отделов лобной и теменной костей может быть сопряжена с выявляемыми при краниографии углублением и расширением на этой же стороне средней черепной ямки, порозностью крыльев основной кости. В таких случаях гемигипертрофия черепа указывает на вероятность наличия в средней черепной ямке неопухолевого объемного процесса (гематома, гигрома, ксантома, кистозный арахноидит и т.п.) и известна как синдром Дайка.
Гемигипертрофия черепа за счет выбухания с одной стороны чешуи височной кости и прилежащих отделов лобной и теменной костей может быть сопряжена с выявляемыми при краниографии углублением и расширением на этой же стороне средней черепной ямки, порозностью крыльев основной кости. В таких случаях гемигипертрофия черепа указывает на вероятность наличия в средней черепной ямке неопухолевого объемного процесса (гематома, гигрома, ксантома, кистозный арахноидит и т.п.) и известна как синдром Дайка.
ПОРОКИ РАЗВИТИЯ ЖЕЛУДОЧКОВ МОЗГА
Пороки развития вентрикулярной системы обычно проявляются в области ее анатомических сужений. Возможны сужения (стеноз и атрезия) межжелудочковых отверстий, водопровода мозга (сильвиева водопровода), срединной и латеральных апертур IV желудочка мозга. В таких случаях характерно развитие внутренней гидроцефалии, при этом в случае атрезии межжелудочкового
отверстия с одной стороны возникает асимметричная гидроцефалия.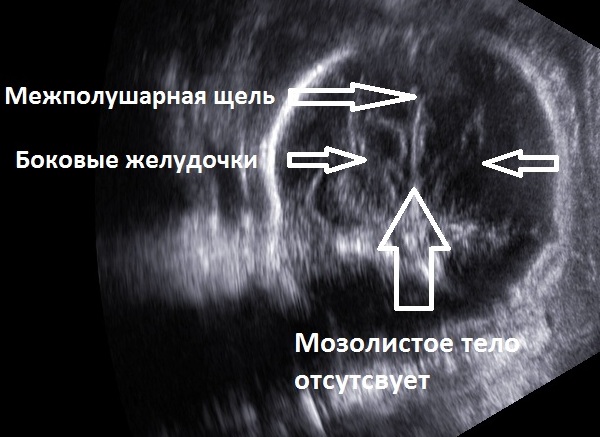 Стеноз или атрезия водопровода мозга, а также его расщепление могут наследоваться, передаваясь по аутосомно-рецессивному типу или быть сцеплены с Х-хромосомой. Неполное раскрытие апертур IV желудочка мозга часто сочетается с проявлениями синдрома Денди-Уокера .
Стеноз или атрезия водопровода мозга, а также его расщепление могут наследоваться, передаваясь по аутосомно-рецессивному типу или быть сцеплены с Х-хромосомой. Неполное раскрытие апертур IV желудочка мозга часто сочетается с проявлениями синдрома Денди-Уокера .
Недостаточность оттока ЦСЖ из желудочковой системы при нарушении проходимости (стенозе) водопровода мозга и апертур IV желудочка мозга проявляется, как правило, развитием внутренней равномерной гидроцефалии, сопровождающейся растяжением, истончением и атрофией ткани мозга. Развитию гидроцефалии нередко сопутствуют и некоторые аномалии основания черепа и верхнего шейного отдела позвоночника: платибазия, симптом Клиппеля- Фейля и др. Возможен также гиперсекреторный или арезорбтивный характер гидроцефалии, обусловленной обычно перенесенным воспалением мозговых оболочек. Частота врожденной гидроцефалии 0,5 на 1000 новорожденных.
ФАКОМАТОЗЫ
Факоматозы (от греч. phakos — пятно, oma — суффикс, означающий «новообразование», «опухоль», osis — суффикс, означающий «процесс», «болезнь») — группа наследственных заболеваний, при которых наблюдается сочетание поражений нервной системы, кожи и внутренних органов. Характерными проявлениями факоматоза являются участки нарушенной пигментации покровных тканей (гиперпигментированные или депигментированные пятна), шагреневые бляшки, фибромы, папилломы, ангиомы, сочетающиеся с разнообразными неврологическими, психическими, эндокринными и соматическими нарушениями. Для большинства форм факоматозов характерны задержки развития различных функций, прежде всего движений и интеллекта, а также снижение адаптации к экзогенным и эндогенным факторам, факторам социальной среды. В тяжелых случаях наблюдаются олигофрения, атаксия, эпилептические припадки. Описания отдельных вариантов факоматоза появились в конце XIX в.
phakos — пятно, oma — суффикс, означающий «новообразование», «опухоль», osis — суффикс, означающий «процесс», «болезнь») — группа наследственных заболеваний, при которых наблюдается сочетание поражений нервной системы, кожи и внутренних органов. Характерными проявлениями факоматоза являются участки нарушенной пигментации покровных тканей (гиперпигментированные или депигментированные пятна), шагреневые бляшки, фибромы, папилломы, ангиомы, сочетающиеся с разнообразными неврологическими, психическими, эндокринными и соматическими нарушениями. Для большинства форм факоматозов характерны задержки развития различных функций, прежде всего движений и интеллекта, а также снижение адаптации к экзогенным и эндогенным факторам, факторам социальной среды. В тяжелых случаях наблюдаются олигофрения, атаксия, эпилептические припадки. Описания отдельных вариантов факоматоза появились в конце XIX в.
Морфологической основой факоматозов являются (Архипов Б. А., Карпухина Л.О., 1996) гамартромы, детерминированные нарушениями роста и дифференцировки клеток одного или нескольких зародышевых листков в ранних стадиях эмбриогенеза. Из клеток, которые как бы задержались в своей дифференцировке и находятся в состоянии «перманентной эмбрионизации», образуются гамартромы, имеющие склонность к пролиферации и неопластической трансформации. В связи с этим гамартрому расценивают как опухолевидный врожденный порок развития или эмбриональную опухоль с бластоматозными тенденциями (Kousseff B.G. et al., 1990). Гамартромы чаще имеют эктодермальное происхождение и состоят из элементов нервной ткани и кожи. Отсюда и другое название факоматозов — «нейроэктодермальные дисплазии». Они могут сочетаться с мезодермальными и энтодермальными дисплазиями.
А., Карпухина Л.О., 1996) гамартромы, детерминированные нарушениями роста и дифференцировки клеток одного или нескольких зародышевых листков в ранних стадиях эмбриогенеза. Из клеток, которые как бы задержались в своей дифференцировке и находятся в состоянии «перманентной эмбрионизации», образуются гамартромы, имеющие склонность к пролиферации и неопластической трансформации. В связи с этим гамартрому расценивают как опухолевидный врожденный порок развития или эмбриональную опухоль с бластоматозными тенденциями (Kousseff B.G. et al., 1990). Гамартромы чаще имеют эктодермальное происхождение и состоят из элементов нервной ткани и кожи. Отсюда и другое название факоматозов — «нейроэктодермальные дисплазии». Они могут сочетаться с мезодермальными и энтодермальными дисплазиями.
Наиболее часто встречаются такие признаки нейроэктодермальной дисплазии, как гипер- и гипопигментированные пятна, пятна цвета «кофе с молоком», фибромы, папилломы, невусы, нейрофибромы, кортикальные и субэпендимальные узелки в ЦНС, факомы, поражения типа «тутовой ягоды» на глазном дне. Среди мезодермальных дисплазий часто встречаются ангиомы, ангиолипомы, аневризмы, эктазии и стенозы сосудов, рабдо- и лейомиомы, дисплазии костной ткани и др. Примером энтодермальной дисплазии может быть полипоз различных отделов пищеварительного тракта.
Среди мезодермальных дисплазий часто встречаются ангиомы, ангиолипомы, аневризмы, эктазии и стенозы сосудов, рабдо- и лейомиомы, дисплазии костной ткани и др. Примером энтодермальной дисплазии может быть полипоз различных отделов пищеварительного тракта.
В каталоге наследственных заболеваний V. McKusik (1967) зарегистрированы 54 формы факоматоза. Большинство из них наследуется по аутосомнодоминантному типу.
Нейрофиброматоз, или болезнь Реклингхаузена, встречается чаще других факоматозов (1:4000). В детском возрасте (после 3 лет) появляются множественные бледные, желто-коричневые (кофейного цвета) пятна, диаметром от просяного зерна до 15 см и больше, преимущественно на туловище и прок- симальных отделах конечностей; нередко наблюдается генерализованная точечная пигментация или веснушчатость в подмышечных областях. Несколько позже появляются признаки нейрофиброматоза: множественные плотные опухоли разной величины (чаще диаметром 1-2 см), расположенные по ходу нервных стволов (невриномы, нейрофибромы), не сращенные с другими тканями.
Опухоли могут возникать и по ходу черепных нервов (невриномы слухового, тройничного, языкоглоточного нервов). Нередко опухоли растут из ткани спинномозговых корешков и располагаются в позвоночном канале, вызывая сдавление спинного мозга. Опухоли могут локализоваться также в глазничной области, в загрудинном, ретроперитонеальном пространствах, во внутренних органах, вызывая соответствующую разнообразную симптоматику. Часто развивается сколиоз, возможна гипертрофия участков кожи, гипертрофия внутренних органов. В основе заболевания лежат аномалии развития экто- и мезодермы. Возможна астроцитарная гамартрома. Наследуется по аутосомно-доминантному типу. Выделяют 2 формы нейрофиброматоза: классическую, периферическую форму (нейрофиброматоз-1), при которой патологический ген локализован в хромосоме 17, и центральную форму (нейрофиброматоз-2), пато- логический ген расположен в хромосоме 22. Описал болезнь в 1882 г. немецкий патолог F.D. Recklinghausen (1833-1910).
Описал болезнь в 1882 г. немецкий патолог F.D. Recklinghausen (1833-1910).
По материалам Института нейрохирургии им. Н.Н. Бурденко РАМН при нейрофиброматозе-1, наряду с периферическими невриномами и нейрофибромами, возможны микроцефалия, пигментированные гамартромы радужки (узелки Лиша), глиомы зрительных нервов (встречаются у 5-10% больных), костные аномалии, в частности дисплазия крыльев основной кости, приводящая к дефекту крыши орбиты и к пульсирующему экзофтальму, односторонние невриномы слухового (вестибулокохлеарного) нерва, внутричерепные опухоли — менингиомы, астроцитомы внутрипозвоночные нейрофибромы, менингиомы, злокачественные опухоли — ганглиобластома, саркома, лейкемия, клинические проявления сирингомиелии.
В случаях нейрофиброматоза-2 часто развивается невринома вестибулокохлеарного черепного нерва, которая при этом заболевании часто бывает двусторонней, возможны менингиома, глиальные опухоли, спинальные невриномы..jpg) Возможны также помутнение хрусталика, субкапсулярная лентикулярная катаракта
Возможны также помутнение хрусталика, субкапсулярная лентикулярная катаракта
Туберозный склероз (болезнь Бурневилля-Прингла, синдром Бурневилля- Брессау) — глиоз белого вещества мозга, проявляющийся в раннем детстве эпилептическими припадками (в 85%), олигофренией в сочетании с нарастающей пирамидной и экстрапирамидной симптоматикой, кожной патологией. В возрасте 4-6 лет на лице в форме бабочки в области носа обычно появляются множественные желто-розовые или коричнево-красные узелки диаметром чуть больше 1 мм — аденомы Прингла, которые обычно признаются аденомами сальных желез, однако есть мнение и о том, что они представляют собой происходящую из нервных элементов кожи гамартрому.
На носу при этом возможны изменения по типу телеангиэктазий. Часто встречаются участки так называемой шагреневой кожи, пятна кофейного цвета, зоны депигментации, полипы, участки фиброзной гиперплазии, возможны гамартромы языка, фиброзные бляшки на коже лба, волосистой части головы и округлые фибромы (опухоли Коэна) на пальцах ног, реже — рук.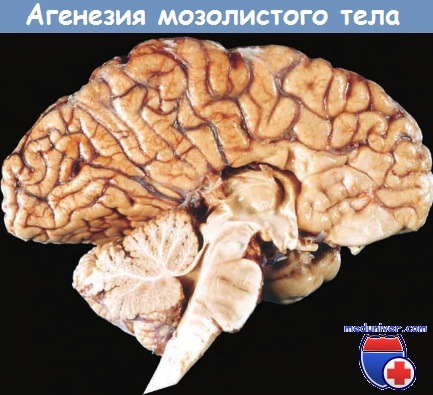 Нередко отмечаются диспластические черты, врожденные пороки развития, опухоли сетчатки и внутренних органов (в сердце, почках, в щитовидной и вилочковой железах и пр.).
Нередко отмечаются диспластические черты, врожденные пороки развития, опухоли сетчатки и внутренних органов (в сердце, почках, в щитовидной и вилочковой железах и пр.).
На глазном дне возможны студенистые образования грязно-желтоватого цвета, напоминающие по форме тутовую ягоду, — глионевромы типа астроцитарной гамартромы, ретинальный факоматоз. Иногда выявляются признаки застоя или атрофии дисков зрительных нервов.
На поверхности мозга наблюдаются единичные или множественные глиоматозные узлы, по цвету несколько светлее окружающего мозга и плотнее его на ощупь, возможна их кальцификация. Узлы могут быть и в белом веществе, подкорковых ганглиях, а также в стволе мозга и в мозжечке.
Встречаются и аномалии развития извилин мозга в виде микро- и пахигирии. Заболевание чаще носит спорадический характер. Бляшки достигают диаметра 5-20 мм. В коре больших полушарий и мозжечка иногда могут быть обнаружены пластинчатые тельца, напоминающие амилоид. Происходит дегенерация клеток коры. При КТ-исследовании головы нередко можно выявить кальцификаты и глиальные узелки в паравентрикулярной области, субэпендимарно вдоль наружных стенок боковых желудочков, в зоне межжелудочкового отверстия Монро, реже — в мозговой паренхиме. На МРТ головного мозга в 60% выявляются гипотеденсивные очаги в одной или обеих затылочных долях, которые расцениваются как участки неправильной миелинизации.
В коре больших полушарий и мозжечка иногда могут быть обнаружены пластинчатые тельца, напоминающие амилоид. Происходит дегенерация клеток коры. При КТ-исследовании головы нередко можно выявить кальцификаты и глиальные узелки в паравентрикулярной области, субэпендимарно вдоль наружных стенок боковых желудочков, в зоне межжелудочкового отверстия Монро, реже — в мозговой паренхиме. На МРТ головного мозга в 60% выявляются гипотеденсивные очаги в одной или обеих затылочных долях, которые расцениваются как участки неправильной миелинизации.
Признается, что заболевание наследуется по аутосомно-доминантному типу с неполной пенетрантностью мутантного гена. Описали его в 1862 г. французский врач D.M. Bourneville (1840-1909) и в 1880 г. английский врач J.J. Pringle (1855-1922).
Энцефалотригеминальный ангиоматоз Стерджа-Вебера (кожно-мозговой ангиоматоз; синдром Штурге (Стерджа)-Вебера; синдром Вебера-Краббе-Ослера — врожденная мальформация мезодермальных (ангиомы) и эктодермальных элементов, возникшая в процессе эмбриогенеза под воздействием экзогенных и генетически обусловленных причин. Характерна триада: «пламенный» невус, эпилепсия, глаукома. Врожденное крупное сосудистое пятно (невус) обычно локализуется на одной стороне лица по ходу ветвей тройничного нерва. Крупные плоские ангиомы красного или вишневого цвета на лице, бледнеющие при надавливании, могут распространяться на кожу волосистой части головы и шею, обычно сопровождаются ангиоматозом мозговых оболочек, чаще в конвекситальной зоне теменно-затылочной области, атрофией мозга и очагами обызвествления в коре больших полушарий. Возможны олигофрения, гемипарез, отставание роста паретичных конечностей, гемианопсия, гидрофтальм. На краниограммах и компьютерных томограммах отмечаются очаги обызвествления, атрофия мозга, расширение субарахноидальных пространств.
Характерна триада: «пламенный» невус, эпилепсия, глаукома. Врожденное крупное сосудистое пятно (невус) обычно локализуется на одной стороне лица по ходу ветвей тройничного нерва. Крупные плоские ангиомы красного или вишневого цвета на лице, бледнеющие при надавливании, могут распространяться на кожу волосистой части головы и шею, обычно сопровождаются ангиоматозом мозговых оболочек, чаще в конвекситальной зоне теменно-затылочной области, атрофией мозга и очагами обызвествления в коре больших полушарий. Возможны олигофрения, гемипарез, отставание роста паретичных конечностей, гемианопсия, гидрофтальм. На краниограммах и компьютерных томограммах отмечаются очаги обызвествления, атрофия мозга, расширение субарахноидальных пространств.
Заболевание чаще носит спорадический характер. Возможны случаи наследования как по доминантному, так и по аутосомно-рецессивному типу. На КТ и МРТ обычно наблюдаются проявления атрофии вещества мозга, расширение желудочков мозга и подоболочечных пространств. Описали заболевание в 1879 г. английские врачи W.H. Sturge (1850-1919) и H.D. Weber (1823-1918).
Описали заболевание в 1879 г. английские врачи W.H. Sturge (1850-1919) и H.D. Weber (1823-1918).
Атаксия-телеангиэктазия (болезнь Луи-Бар) характеризуется симметричными телеангиэктазиями, появляющимися в возрасте 3-6 лет, особенно на конъюнктиве, коже лица и шеи, обычно распространяющимися на мозговые оболочки, вещество мозга. Кроме того, отмечается повышенная склонность к хроническим воспалительным заболеваниям (синусит, пневмония, бронхоэктазии и пр.) в связи с генетически обусловленным нарушением клеточного и гуморального иммунитета. При первых попытках ребенка к самостоятельной ходьбе выявляются признаки мозжечковой атаксии, которая в дальнейшем имеет нарастающий характер, позднее появляются гиперкинезы по типу миоклонии или атетоза, сухожильная гипорефлексия, дизартрия. Возможно поражение черепных нервов, затруднение произвольных движений глаз (окуломоторная апраксия) .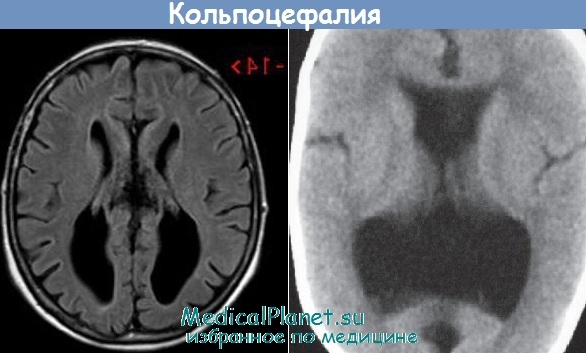 К 12-15 годам возникают нарушения глубокой и вибрационной чувствительности, нарастание атаксии. В поздних стадиях болезни в связи с поражением клеток передних рогов спинного мозга возникают слабость и атрофия мышц, фасцикулярные подергивания. На коже появляются пигментные пятна кофейного цвета, участки гипопигментации, себорейный дерматит. Постепенно развивается атрофия кожи, появление седых волос отмечается уже в школьном возрасте. Характерны задержка психического и физического развития, обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммаглобулинемия, поражение ретикулоэндотелиальной системы (ретикулезы, лимфосаркома и пр.). Прогноз плохой. Причиной смерти чаще являются хронические заболевания бронхов и легких, лимфомы, карциномы.
К 12-15 годам возникают нарушения глубокой и вибрационной чувствительности, нарастание атаксии. В поздних стадиях болезни в связи с поражением клеток передних рогов спинного мозга возникают слабость и атрофия мышц, фасцикулярные подергивания. На коже появляются пигментные пятна кофейного цвета, участки гипопигментации, себорейный дерматит. Постепенно развивается атрофия кожи, появление седых волос отмечается уже в школьном возрасте. Характерны задержка психического и физического развития, обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммаглобулинемия, поражение ретикулоэндотелиальной системы (ретикулезы, лимфосаркома и пр.). Прогноз плохой. Причиной смерти чаще являются хронические заболевания бронхов и легких, лимфомы, карциномы.
Наследуется по аутосомно-рецессивному типу с высокой пенетрантностью мутантного гена. Описала болезнь в 1941 г. французская врач D. Louis-Bar.
Цереброретиновисцеральный ангиоматоз (гемангиобластоматоз, болезнь Гиппеля-Линдау) — наследственно-семейный ангиоматоз центральной нервной системы и сетчатки глаз. Характеризуется врожденным недоразвитием капилляров, компенсаторным расширением более крупных сосудов и образованием сосудистых клубочков, ангиом, ангиоглиом. Неврологическая симптоматика может быть разнообразной в связи с возможным поражением больших полушарий, ствола мозга, мозжечка, реже — спинного мозга.
Характеризуется врожденным недоразвитием капилляров, компенсаторным расширением более крупных сосудов и образованием сосудистых клубочков, ангиом, ангиоглиом. Неврологическая симптоматика может быть разнообразной в связи с возможным поражением больших полушарий, ствола мозга, мозжечка, реже — спинного мозга.
Характерна триада: ангиома сетчатки, ангиомы головного мозга, поликистоз внутренних органов или ангиоретикулема почек. На глазном дне отмечаются резкое расширение и извитость сосудов, желтоватые сосудистые клубочки в сетчатке, позже — экссудат и кровоизлияния в сетчатке, ее отслойка. Часто наблюдаются помутнение стекловидного тела, глаукома, иридоциклит. В результате со временем наступает слепота. Болезнь Гиппеля-Линдау обычно проявляется у больных 18-50 лет.
Первыми симптомами являются признаки ангиоретикулемы мозжечка или сетчатки глаз. При преобладании клинических проявлений ангиоматоза мозжечка болезнь известна как «опухоль Линдау». Ангиоматоз сетчатки обычно рассматривается как «опухоль Гиппеля». Возможны поражения внутренних органов, которые характеризуются аномалиями развития и образованием опухолей: поликистоза почек, феохромоцитомы, гипернефромы, кистозных опухолей поджелудочной железы, печени. Наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Описали болезнь в 1904 г. немецкий офтальмолог Е. Hippel, а в 1925 г. шведский патолог А. Lindau (род. в 1898 г.).
Ангиоматоз сетчатки обычно рассматривается как «опухоль Гиппеля». Возможны поражения внутренних органов, которые характеризуются аномалиями развития и образованием опухолей: поликистоза почек, феохромоцитомы, гипернефромы, кистозных опухолей поджелудочной железы, печени. Наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Описали болезнь в 1904 г. немецкий офтальмолог Е. Hippel, а в 1925 г. шведский патолог А. Lindau (род. в 1898 г.).
АНОМАЛИИ И ДЕСТРУКЦИИ НА КРАНИОВЕРТЕБРАЛЬНОМ УРОВНЕ
В зоне перехода черепа в позвоночник часто встречаются краниовертебральные аномалии. Они могут обусловить нарушение кровообращения в позвоночных артериях, расстройство ликворообращения. В результате проявления разнообразных неврологических расстройств, в том числе вестибулярные, мозжечковые симптомы, признаки внутричерепной гипертензии, элементы бульбарного синдрома, в частности нарушения функций черепных нервов бульбарной группы, корешковые симптомы на верхнешейном уровне, признаки пирамидной недостаточности, нарушения чувствительности по проводниковому типу, а также корешковые симптомы на верхнешейном уровне. Могут выявляться многообразные костные аномалии, проявления дизрафического статуса: базилярное вдавление, выстояние верхушки зубовидного отростка выше линий Чемберлена и де ля Пети, ассимиляция атланта (синдром Ольенека), феномен проатланта и др. Для краниовертебральных аномалий характерны короткая шея, низкая граница роста волос на шее, шейный гиперлордоз; возможны асимметрия лица, гипоплазия нижней челюсти, готическое нёбо, расширение позвоночного канала на уровне верхних шейных позвонков, кифосколиоз позвоночника, расщепление дужек позвонков, деформация стоп по типу «стопы Фридрейха».
Могут выявляться многообразные костные аномалии, проявления дизрафического статуса: базилярное вдавление, выстояние верхушки зубовидного отростка выше линий Чемберлена и де ля Пети, ассимиляция атланта (синдром Ольенека), феномен проатланта и др. Для краниовертебральных аномалий характерны короткая шея, низкая граница роста волос на шее, шейный гиперлордоз; возможны асимметрия лица, гипоплазия нижней челюсти, готическое нёбо, расширение позвоночного канала на уровне верхних шейных позвонков, кифосколиоз позвоночника, расщепление дужек позвонков, деформация стоп по типу «стопы Фридрейха».
Врожденные аномалии развития на краниовертебральном уровне характеризуются дефектами развития затылочной кости и расположенных в задней черепной ямке структур и верхнего отдела позвоночника и спинного мозга. К ним относятся синдромы Денди-Уокера и синдром Киари.
Синдром Денди-Уокера представляет собой врожденный порок развития каудального отдела ствола и червя мозжечка, ведущий к неполному раскрытию срединной (Мажанди) и латеральных (Лушки) апертур IV желудочка мозга. Проявляется признаками гидроцефалии, а нередко и гидромиелии. Последнее обстоятельство в соответствии с гидродинамической теорией Гарднера может обусловить развитие сирингомиелии, сирингобульбии. Синдром Денди-Уокера характеризуется проявлениями функциональной недостаточности продолговатого мозга и мозжечка, симптомами гидроцефалии, внутричерепной гипертензии. Уточняется диагноз с помощью визуализирующих мозговую ткань методов — КТ- и МРТ-исследований. Выявляются признаки гидроцефалии, в частности выраженное расширение IV желудочка мозга, при МРТ-исследовании можно выявить деформацию указанных структур мозга. Описали синдром в 1921 г. американские нейрохирурги W. Dandy (1886-1946) и A. Walker (род. в 1907 г.).
Проявляется признаками гидроцефалии, а нередко и гидромиелии. Последнее обстоятельство в соответствии с гидродинамической теорией Гарднера может обусловить развитие сирингомиелии, сирингобульбии. Синдром Денди-Уокера характеризуется проявлениями функциональной недостаточности продолговатого мозга и мозжечка, симптомами гидроцефалии, внутричерепной гипертензии. Уточняется диагноз с помощью визуализирующих мозговую ткань методов — КТ- и МРТ-исследований. Выявляются признаки гидроцефалии, в частности выраженное расширение IV желудочка мозга, при МРТ-исследовании можно выявить деформацию указанных структур мозга. Описали синдром в 1921 г. американские нейрохирурги W. Dandy (1886-1946) и A. Walker (род. в 1907 г.).
Синдром Киари (синдром Арнольда-Киари-Соловцева, или синдром церебелломедулярного уродства) — порок развития субтенториальных структур ромбовидного мозга, проявляющийся опущением ствола мозга и миндалин мозжечка в большое затылочное отверстие..png) Нередко сочетается с аномалиями костей основания черепа и верхних шейных позвонков (платибазией, базилярной импрессией, ассимиляцией атланта, синдромом Клиппеля-Фейля), с проявлениями дизрафического статуса, в частности с сирингомиелией, сирингобульбией. При синдроме Киари могут возникать ущемление продолговатого мозга, структур мозжечка, верхних шейных сегментов спинного мозга, окклюзия ликворных путей, что ведет к бульбарным, мозжечковым и проводниковым симптомам, к окклюзионной гидроцефалии. Описали синдром в 1894 г. немецкий патолог J. Arnold (1835-1915) и в 1895 г. австрийский патолог H. Chiari (1851-1916).
Нередко сочетается с аномалиями костей основания черепа и верхних шейных позвонков (платибазией, базилярной импрессией, ассимиляцией атланта, синдромом Клиппеля-Фейля), с проявлениями дизрафического статуса, в частности с сирингомиелией, сирингобульбией. При синдроме Киари могут возникать ущемление продолговатого мозга, структур мозжечка, верхних шейных сегментов спинного мозга, окклюзия ликворных путей, что ведет к бульбарным, мозжечковым и проводниковым симптомам, к окклюзионной гидроцефалии. Описали синдром в 1894 г. немецкий патолог J. Arnold (1835-1915) и в 1895 г. австрийский патолог H. Chiari (1851-1916).
В настоящее время, основываясь на результатах МРТ-сканирования, некоторые авторы выделяют два варианта синдрома Киари.
Мальформация I типа (Киари I) характеризуется смещением миндалин мозжечка до уровня большого затылочного отверстия. Возможно опущение продолговатого мозга, его удлинение и передняя компрессия продолговатого мозга зубовидным отростком, сужение IV желудочка мозга и большой затылочной цистерны, ликвородинамические расстройства, признаки недоразвития и атипичного строения артерий вертебрально-базилярного бассейна..jpg) В неврологическом статусе возможны глазодвигательные, кохлеарные и вестибуломозжечковые, бульбарные, а также проводниковые двигательные и сегментарные двигательные и чувствительные нарушения. Отсутствие неврологических симптомов, однако они могут проявиться позже (иногда на 3-4 десятилетии жизни, что свидетельствует о переходе процесса в мальформацию II типа.
В неврологическом статусе возможны глазодвигательные, кохлеарные и вестибуломозжечковые, бульбарные, а также проводниковые двигательные и сегментарные двигательные и чувствительные нарушения. Отсутствие неврологических симптомов, однако они могут проявиться позже (иногда на 3-4 десятилетии жизни, что свидетельствует о переходе процесса в мальформацию II типа.
При мальформации II типа (Киари II) отмечается протрузия в большое затылочное отверстие миндалин и червя мозжечка, структур продолговатого мозга, который принимает S-образную форму. Характерны спастический тетрапарез, боли в затылочной области и шее, мозжечковая атаксия, вертикальный «бьющий» вниз нистагм, элементы бульбарного синдрома, признаки сирингомиелии, проявления гидроцефалии, проводниковые нарушения.
Неврологическая симптоматика при синдроме Арнольда-Киари может проявляться с 5-7 лет, иногда позже, возможно в 30-40-летнем возрасте, и имеет прогредиентное течение. Проявления аномалии Арнольда-Киари нередко сочетаются с краниовертебральной костной аномалией (базилярная импрессия, ассимиляция атланта, краниостеноз по типу скафокрании и др. ). В диагностике синдрома Киари и определении его типа особенно ценной обычно является информация, полученная при МРТ головного мозга и краниовертебральной области, а также при транскраниальной допплерографии (Крупина Н.Е., 2003).
). В диагностике синдрома Киари и определении его типа особенно ценной обычно является информация, полученная при МРТ головного мозга и краниовертебральной области, а также при транскраниальной допплерографии (Крупина Н.Е., 2003).
Симптом Бабчина — атрофия заднего полукольца большого затылочного отверстия и внутреннего гребешка затылочной кости. Выявляется при краниографии, выполненной в задней полуаксиальной проекции. Описан симптом отечественным нейрохирургом И.С. Бабчиным при опухолях краниовертебральной локализации.
НЕКОТОРЫЕ ВРОЖДЕННЫЕ ИЛИ РАНО ПРОЯВЛЯЮЩИЕСЯ ФОРМЫ ПОРАЖЕНИЯ ДВИГАТЕЛЬНОЙ СФЕРЫ
Детские церебральные параличи
Детские церебральные параличи (ДЦП) — гетерогенная группа синдромов, которые являются следствием повреждений мозга, возникших во внутриутробном, интранатальном (во время родов) и раннем постнатальном периодах. Характерная особенность ДЦП — нарушение моторного развития ребенка, обусловленное прежде всего аномальным распределением мышечного тонуса и нарушением координации движений (парезы, параличи, атаксия, гиперкинезы). Отмеченные двигательные расстройства могут сочетаться с приступами эпилепсии, задержкой развития речи, эмоционального и интеллектуального развития. Иногда расстройства движений сопровождаются и изменением чувствительности.
Отмеченные двигательные расстройства могут сочетаться с приступами эпилепсии, задержкой развития речи, эмоционального и интеллектуального развития. Иногда расстройства движений сопровождаются и изменением чувствительности.
Важной особенностью ДЦП является отсутствие прогрессирования и возможная, хотя и слабо выраженная, тенденция к восстановлению имеющихся признаков патологии нервной системы.
Частота ДЦП, по разным данным, составляет 2,5-5,9 на 1000 новорожденных. По данным Московской детской консультативной неврологической поликлиники, в 1977-1978 гг. она составила 3,3 на 1000 детского населения. Частота ДЦП в группе детей, родившихся с массой тела менее 1500 г, составляет 5-15% (Aziz K. et al., 1994). Согласно данным К.А. Семеновой (1994), ДЦП является причиной 24% случаев детской неврологической инвалидности.
Этиология. Этиологические факторы разнообразны: заболевания (краснуха, цитомегалия, грипп, токсоплазмоз и др.) и токсикозы у матери во время беременности, аномалии родовой деятельности, акушерские операции и травматические поражения, кровоизлияния в мозг, асфиксии во время родового акта, несовместимость крови матери и плода, травмы и болезни (менингиты, энцефалиты) у ребенка в раннем послеродовом периоде..jpg) Возможно сочетание нескольких вредных факторов.
Возможно сочетание нескольких вредных факторов.
Причинами врожденного ДЦП могут быть генетические детерминированные аномалии формирования мозга (дисгенезии мозга), возникающие на разных этапах его развития. Они являются причиной 10-11% всех случаев спастических форм ДЦП. Кроме того, причиной ДЦП могут быть сосудисто-мозговые нарушения у плода или новорожденного ребенка, в частности гипоксически-ишемическая энцефалопатия, ишемические и геморрагические инсульты, внутричерепные гематомы.
Патогенез. Патогенные факторы, действующие во время эмбригенеза, вызывают аномалии развития мозга. На более поздних этапах внутриутробного развития возможно замедление процессов миелинизации нервной системы, нарушение дифференциации нервных клеток, патология формирования межнейронных связей и сосудистой системы мозга. При несовместимости крови матери и плода по резус-фактору, системе АВ0 и другим антигенам эритроцитов в организме матери вырабатываются антитела, вызывающие гемолиз эритроцитов плода.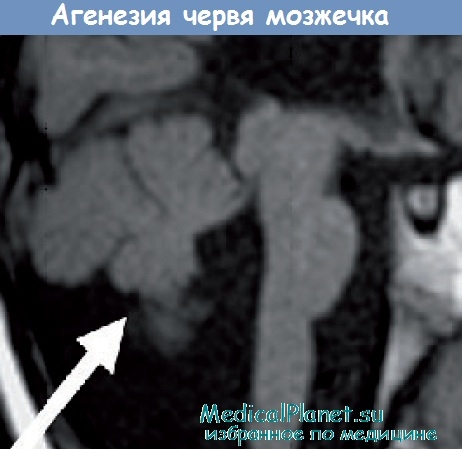 Непрямой билирубин, образовавшийся в процессе гемолиза, оказывает токсическое действие на нервную систему, в частности на структуры стриопаллидарной системы.
Непрямой билирубин, образовавшийся в процессе гемолиза, оказывает токсическое действие на нервную систему, в частности на структуры стриопаллидарной системы.
У плодов, перенесших внутриутробную гипоксию, к моменту рождения защитные и адаптационные механизмы оказываются недостаточно сформированными, существенное значение могут иметь асфиксия и черепно-мозговая травма во время родов. В патогенезе поражений нервной системы, развивающихся во время родов и постнатально, главную роль играют гипоксия плода, ацидоз, гипогликемия и другие метаболические нарушения, ведущие к отеку мозга и вторичным расстройствам мозговой гемодинамики и ликвородинамики. Существенное значение в патогенезе ДЦП придается иммунопатологическим процессам: мозговые антигены, образующиеся при деструкции нервной системы под влиянием инфекций, интоксикаций, механических повреждений мозговой ткани, могут привести к появлению соответствующих антител в крови матери, что негативно сказывается на развитии мозга плода.
Патоморфологическая картина. Патоморфологические изменения нервной системы при ДЦП многообразны. У 30% детей имеются аномалии развития мозга — микрогирия, пахигирия, гетеротопия, недоразвитие полушарий и др. Возможны дистрофические изменения мозга, глиоматоз, рубцы, порэнцефалия или кистозные полости в мозге, участки демиелинизации проводящих путей или атрофии коры больших полушарий в связи с травматическим поражением, кровоизлиянием в мозг, внутричерепной гематомой, гипоксией, возникшими в процессе родового акта или токсическим, инфекционно-аллергическим, травматическим поражением мозга во внутриутробном или раннем постнатальном периодах.
Классификация. Предлагаются разные клинические классификации ДЦП. Мы приводим одну из классификаций, получивших широкое признание.
Таблица 24.1. Синдромы (формы) ДЦП (Miller G., 1998)
Преобладающими являются спастические формы, остальные встречаются значительно реже.
Клинические проявления. Возникший дефект в мозге не только негативно сказывается на состоянии новорожденного ребенка, но и препятствует его нормальному развитию, прежде всего развитию двигательной системы, речи и когнитивных функций. Клиническая картина в таких случаях может варьировать в больших пределах. Важно помнить, что патологическая постуральная активность, проявления повышения мышечного тонуса нередко становятся отчетливыми только к 3-4 месяцу жизни ребенка, а иногда и позже. Для относительно ранней диагностики ДЦП имеет значение динамическое наблюдение за детьми, особенно с неблагополучным акушерским анамнезом, учет при этом динамики врожденных безусловных рефлексов, последовательности характера изменений мышечного тонуса становления реакций выпрямления и равновесия.
Возникший дефект в мозге не только негативно сказывается на состоянии новорожденного ребенка, но и препятствует его нормальному развитию, прежде всего развитию двигательной системы, речи и когнитивных функций. Клиническая картина в таких случаях может варьировать в больших пределах. Важно помнить, что патологическая постуральная активность, проявления повышения мышечного тонуса нередко становятся отчетливыми только к 3-4 месяцу жизни ребенка, а иногда и позже. Для относительно ранней диагностики ДЦП имеет значение динамическое наблюдение за детьми, особенно с неблагополучным акушерским анамнезом, учет при этом динамики врожденных безусловных рефлексов, последовательности характера изменений мышечного тонуса становления реакций выпрямления и равновесия.
По преобладанию тех или иных неврологических и психических функций Л.О. Бадалян (1984) выделял следующие варианты ДЦП.
1. Спастическая диплегия (синдром Литтля) — наиболее часто встречающаяся форма ДЦП. Характеризуется тетрапарезом с вовлечением в процесс мышц лица, языка, глотки, при этом особенно выражены двигательные расстройства в нижних конечностях (проявления нижнего спастического парапареза с преобладанием напряжения приводящих мышц бедер и мышц-разгибателей голени и сгибателей стоп. Если ребенок лежит, ноги у него вытянуты, при попытке поставить на пол) ноги у него перекрещиваются, он опирается не на всю стопу, а только на переднюю ее часть. Ноги при этом выпрямлены и ротированы внутрь. При попытке ходить с посторонней помощью ребенок совершает танцующие движения, ноги его «перекрещиваются», тело поворачивается в сторону ведущей ноги. Нередко выраженность парезов асимметрична, при этом различие в возможности активных движений особенно отчетливо проявляется в руках.
Характеризуется тетрапарезом с вовлечением в процесс мышц лица, языка, глотки, при этом особенно выражены двигательные расстройства в нижних конечностях (проявления нижнего спастического парапареза с преобладанием напряжения приводящих мышц бедер и мышц-разгибателей голени и сгибателей стоп. Если ребенок лежит, ноги у него вытянуты, при попытке поставить на пол) ноги у него перекрещиваются, он опирается не на всю стопу, а только на переднюю ее часть. Ноги при этом выпрямлены и ротированы внутрь. При попытке ходить с посторонней помощью ребенок совершает танцующие движения, ноги его «перекрещиваются», тело поворачивается в сторону ведущей ноги. Нередко выраженность парезов асимметрична, при этом различие в возможности активных движений особенно отчетливо проявляется в руках.
На фоне диплегии могут быть хореоатетоидные гиперкинезы, в которые вовлекаются прежде всего мимические мышцы и мышцы дистальных отделов рук. Дети тяжело переживают наличие двигательных нарушений, неохотно вступают в контакт со здоровыми детьми, лучше чувствуют себя в коллективе, состоящем из детей с подобными болезнями.
2. Двойная гемиплегия — двусторонняя гемиплегия или, чаще, гемипарез, при котором руки страдают в большей степени, чем ноги, или они поражены приблизительно в равной мере. Возможна асимметрия выраженности парезов, при этом тонус мышц высокий, имеется сочетание спастики и ригидности обычно с преобладанием последней. Реакции равновесия развиты недостаточно. Почти всегда выражены элементы псевдобульбарного паралича, в связи с чем затруднены жевание и глотание, речь. Нередко отмечаются судорожные пароксизмы, микроцефалия. Эта форма ДЦП обычно сопровождается наиболее значительными проявлениями олигофрении.
3. Спастическая гемиплегия характеризуется соответствующими двигательными нарушениями преимущественно на одной стороне. Нередко двигательные расстройства более выражены в руке, она согнута во всех суставах, кисть у детей раннего возраста сжата в кулак, в более позднем возрасте имеет форму «руки акушера». Нередко возникают фокальные эпилептические припадки по типу Джексона.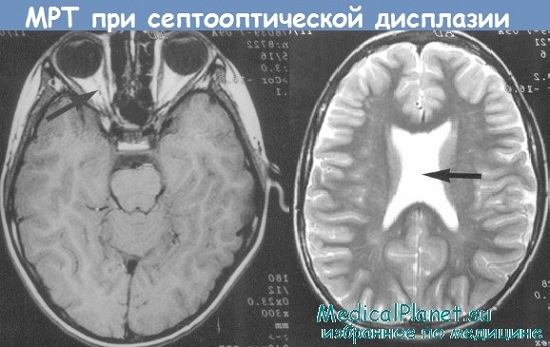 С помощью визуализирующих методов исследования (КТ, МРТ) в одном из полушарий мозга обычно выявляют кисту, рубцовые процессы или проявления порэнцефалии. Развитие интеллекта может оказаться близким к нормальному.
С помощью визуализирующих методов исследования (КТ, МРТ) в одном из полушарий мозга обычно выявляют кисту, рубцовые процессы или проявления порэнцефалии. Развитие интеллекта может оказаться близким к нормальному.
4. Гиперкинетическая форма характеризуется преимущественным поражением структур стриопаллидарной системы. Мышечный тонус изменчив, часто колеблется между гипотонией и нормотонией. На этом фоне возникают перемежающиеся мышечные спазмы, приступы повышения мышечного тонуса по пластическому типу. Активные движения в таких случаях неловкие, сопровож- даются излишними двигательными реакциями преимущественно атетоидного характера, при этом гиперкинезы могут быть преимущественно в дистальных или проксимальных отделах конечностей, мышцах шеи, в мимической мускулатуре. Гиперкинезы возможны по типу атетоза, хореоатетоза, хореи, торсионной дистонии. Часто наблюдаются расстройства речи (подкорковая дизартрия). Психическое развитие страдает меньше, чем при других формах ДЦП.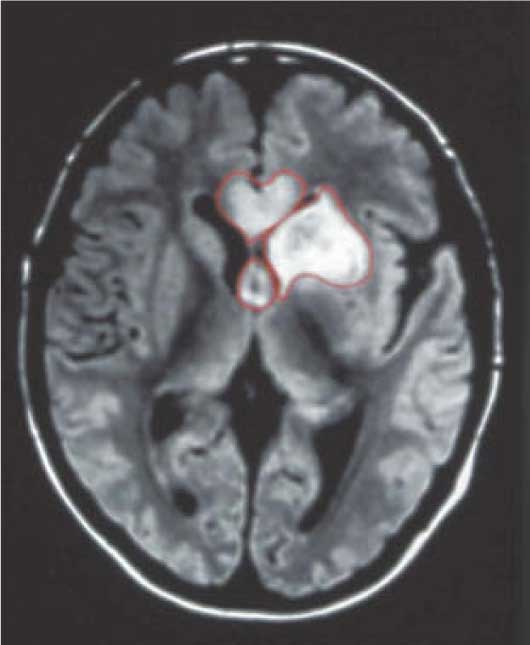 Эта форма ДЦП обычно обусловлена иммунной несовместимостью крови плода и матери.
Эта форма ДЦП обычно обусловлена иммунной несовместимостью крови плода и матери.
5. Мозжечковая форма характеризуется атаксией, обусловленной главным образом поражением мозжечка и его связей. Может сочетаться с нистагмом, атонически-астатическим синдромом, признаками умеренного спастического пареза в связи с вовлечением в процесс корково-подкорковых структур мозга.
Лечение. Лечение, точнее, абилитацию1 больного с ДЦП следует начинать как можно раньше, при этом оно должно быть комплексным. В раннем возрасте мозг ребенка пластичен и обладает значительными компенсаторными возможностями. Абилитация, начатая в период формирования статических и локомоторных функций, дает наиболее существенные результаты. Раннее обучение сенсомоторным навыкам с условнорефлекторным их закреплением способствует своевременному развитию моторики. Кроме того, в раннем возрасте спастические явления выражены еще нерезко, отсутствуют стереотипные патологические позы, деформации, контрактуры, вследствие чего легче вырабатываются двигательные навыки.![]()
1Абилитация — создание возможностей для развития отсутствовавших ранее видов деятельности.
Важной частью комплексного лечения ДЦП являются ортопедические мероприятия, профилактика контрактур. Для придания физиологического положения отдельным частям тела широко используются лонгеты, туторы, шины, валики, воротники и пр. Ортопедические укладки чередуются с лечебной гимнастикой, массажем, физиотерапией, при этом лечебные мероприятия должны способствовать торможению патологической тонической рефлекторной активности, нормализации на этой основе мышечного тонуса, облегчению произвольных движений, развитию последовательных возрастных двигательных навыков ребенка.
Из лекарственных средств в процессе лечения ДЦП применяются фармакопрепараты, улучшающие метаболические процессы в мозге, — глутаминовая кислота, липоцеребрин, церебролизин, препараты из группы ноотропов, витамины группы В, ацефен и др. По показаниям применяют миорелаксанты, при этом средством выбора может быть ботокс (ботулинический токсин).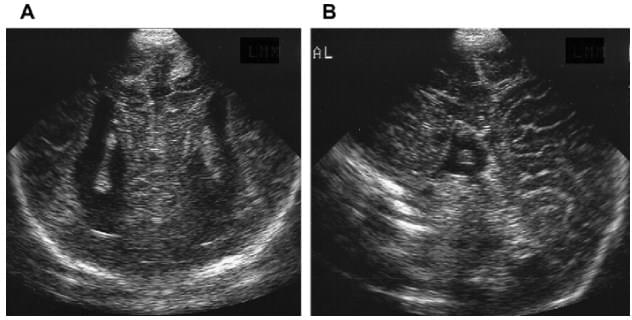 Есть положительный опыт (Белоусова Е.Д., Темин П.А. и др., 1999) его введения в двуглавую мышцу плеча, а также в сгибатели и разгибатели кисти для уменьшения мышечного тонуса и пронаторной установки предплечья; положительный эффект дало применение ботокса теми же авторами для ликвидации динамической контрактуры в голеностопном суставе. Применяются также препараты, действие которых направлено на подавление гиперкинезов, противосудорожные средства, ангиопротекторы, антиагреганты и седативные средства.
Есть положительный опыт (Белоусова Е.Д., Темин П.А. и др., 1999) его введения в двуглавую мышцу плеча, а также в сгибатели и разгибатели кисти для уменьшения мышечного тонуса и пронаторной установки предплечья; положительный эффект дало применение ботокса теми же авторами для ликвидации динамической контрактуры в голеностопном суставе. Применяются также препараты, действие которых направлено на подавление гиперкинезов, противосудорожные средства, ангиопротекторы, антиагреганты и седативные средства.
В последние годы разрабатываются методы соматосенсорной стимуляции. Для этого предлагается, в частности, ношение космического нагрузочного костюма «Пингвин» или его модификации «Адели». Применение нагрузочного костюма способствует исправлению положения центра тяжести тела больного и нормализации позы стояния. Предполагается (Яворский А.Б. и др., 1998), что при таком лечении может происходить перестройка нервных связей в полушариях мозга и изменение при этом межполушарных взаимоотношений..jpg)
Спастическая семейная параплегия Штрюмпелля
Хроническая прогрессирующая семейная болезнь подробно описана в 1886 г. немецким врачом А. Штрюмпеллем (Stumpell A., 1853-1925). В настоящее время она расценивается как группа заболеваний, характеризующаяся генетической гетерогенностью и клиническим полиморфизмом. Болезнь наследуется как по аутосомно-рецессивному, так и по доминантному типу.
Патогенез не изучен.
Патоморфологическая картина. Отмечается симметричная дегенерация в церебро-спинальных проводящих путях, постепенно прогрессирующая и распространяющаяся снизу вверх. Иногда она сопровождается дегенеративными изменениями в нежном пучке Голля и спинномозговых трактах. Возможны демиелинизация нервных волокон в ножках мозга, глиоз и уменьшение количества клеток в экстрапирамидных структурах ствола.
Клинические проявления. Обычно на втором десятилетии жизни появляются утомляемость ног, повышение в них мышечного тонуса и сухожильных рефлексов. Позже возникают клонусы стоп, стопные патологические знаки. Со временем признаки нижнего спастического парапареза нарастают, при этом спастическое состояние мышц преобладает над выраженностью мышечной
Позже возникают клонусы стоп, стопные патологические знаки. Со временем признаки нижнего спастического парапареза нарастают, при этом спастическое состояние мышц преобладает над выраженностью мышечной
слабости. В течение многих лет больные сохраняют способность к самостоятельному передвижению. Походка у них спастическая парапаретическая. Изза выраженности напряжения приводящих мышц бедер больные иногда при ходьбе перекрещивают ноги. В далеко зашедшей стадии заболевания возможны защитные рефлексы, признаки спинномозгового автоматизма, контрактуры голеностопных суставов. Элементы спастичности могут по мере развития заболевания проявиться в руках, в мышцах плечевого пояса. Возможно снижение вибрационной чувствительности на ногах. Другие виды чувствительности, трофика тканей и функции тазовых органов обычно не страдают. Возможны деформация стоп (стопа Фридрейха), легкая мозжечковая недостаточность, миокардиопатия, снижение когнитивных функций.
Лечение. Патогенетическая терапия не разработана. В качестве симптоматических средств широко применяются миорелаксанты (мидокалм, скутамил, баклофен и др.).
В качестве симптоматических средств широко применяются миорелаксанты (мидокалм, скутамил, баклофен и др.).
Особенности высших психических функций при патологии мозолистого тела
Проанализированы публикации, посвященные вкладу мозолистого тела (МТ) в организацию высших психических функций (ВПФ) и в межполушарное взаимодействие. Большинство авторов приходят к выводу, что при нейропсихологическом обследовании лиц с патологией МТ можно выявить проблемы в различных ВПФ. Изложены результаты нейропсихологического обследования 29 лиц с различными вариантами патологии МТ.
Higher mental functions in callosal pathology .pdf Проблема межполушарного взаимодействия — одна из наиболее актуальных проблем современной нейропсихологии. Сегодня идет интенсивное накопление и осмысление клинических и экспериментальных данных о механизмах, лежащих в основе межполушарного взаимодействия. Именно наличие функциональных связей между полушариями обеспечивает преимущества мозга как парного органа. Хотя нейропсихологи давно говорят о существовании фактора межполушарного взаимодействия, это понятие пока недостаточно наполнено конкретным содержанием [1].Анатомическим субстратом межполушарного взаимодействия являются многочисленные мозговые ко-миссуры. Ведущая роль принадлежит самой крупной комиссуре — мозолистому телу. Раскрытие нейропси-хологических механизмов функционирования комис-сурной системы мозга, ее вклада в реализацию любой психической функции является важной задачей современной нейропсихологии.Изучение мозга детей и взрослых показало, что миелинизация передней комиссуры и МТ заканчивается к подростковому возрасту [2]. Современные МРТ-исследования обнаружили, что МТ созревает к концу второго десятилетия жизни [3-5]. По другим данным, размер МТ продолжает увеличиваться до середины двадцатилетия [6]. Значительное увеличение размера МТ выявлено у детей между 4 и 18 годами, причем увеличение МТ в детском возрасте, скорее всего, связано с миелинизацией, а не с увеличением количества аксонов, что косвенно подтверждается данными, полученными при изучении приматов [7, 8].
Хотя нейропсихологи давно говорят о существовании фактора межполушарного взаимодействия, это понятие пока недостаточно наполнено конкретным содержанием [1].Анатомическим субстратом межполушарного взаимодействия являются многочисленные мозговые ко-миссуры. Ведущая роль принадлежит самой крупной комиссуре — мозолистому телу. Раскрытие нейропси-хологических механизмов функционирования комис-сурной системы мозга, ее вклада в реализацию любой психической функции является важной задачей современной нейропсихологии.Изучение мозга детей и взрослых показало, что миелинизация передней комиссуры и МТ заканчивается к подростковому возрасту [2]. Современные МРТ-исследования обнаружили, что МТ созревает к концу второго десятилетия жизни [3-5]. По другим данным, размер МТ продолжает увеличиваться до середины двадцатилетия [6]. Значительное увеличение размера МТ выявлено у детей между 4 и 18 годами, причем увеличение МТ в детском возрасте, скорее всего, связано с миелинизацией, а не с увеличением количества аксонов, что косвенно подтверждается данными, полученными при изучении приматов [7, 8]. Экспериментальные исследования показали, что через МТ передается уже первично обработанная во вторичных и третичных корковых полях сенсорная информация [9-11]. Роль МТ невелика в переносе моторных команд, но она возрастает при переносе латерализованной вербальной или зрительно-пространственной информации [12]. Межполушарное взаимодействие возрастает при повышении трудности выполняемого задания [13-15]. Одна из важных функций МТ — обеспечение возможности межполушарного торможения для дифференциации активности полушарий и более эффективной обработки информации. От качества межполушарного взаимодействия зависит и уровень интеллекта [16].Интерес к изучению функций МТ резко усилился под влиянием исследований пациентов с каллозотоми-ей. Это позволило, с одной стороны, верифицировать и дополнить сложившиеся представления о функциях правого и левого полушарий мозга; с другой — оценить функциональное значение комиссурной системы мозга. Результаты психологических экспериментов и наблюдений показали, что разделение полушарий приводит к возникновению двух видов мозга «в одном черепе» [17.
Экспериментальные исследования показали, что через МТ передается уже первично обработанная во вторичных и третичных корковых полях сенсорная информация [9-11]. Роль МТ невелика в переносе моторных команд, но она возрастает при переносе латерализованной вербальной или зрительно-пространственной информации [12]. Межполушарное взаимодействие возрастает при повышении трудности выполняемого задания [13-15]. Одна из важных функций МТ — обеспечение возможности межполушарного торможения для дифференциации активности полушарий и более эффективной обработки информации. От качества межполушарного взаимодействия зависит и уровень интеллекта [16].Интерес к изучению функций МТ резко усилился под влиянием исследований пациентов с каллозотоми-ей. Это позволило, с одной стороны, верифицировать и дополнить сложившиеся представления о функциях правого и левого полушарий мозга; с другой — оценить функциональное значение комиссурной системы мозга. Результаты психологических экспериментов и наблюдений показали, что разделение полушарий приводит к возникновению двух видов мозга «в одном черепе» [17. С. 132]. После операции по «рассечению мозга» у больных не наблюдается отчетливых изменений темперамента, личности, общего интеллекта. Кроме того, описанный синдром является нестойким — со временем, в течение нескольких недель, выраженность симптомов существенно уменьшается, а иногда они вообще исчезают [1,18].Отечественные нейропсихологи исследовали больных с частичным рассечением МТ в результате удаления артериовенозных аневризм. Было установлено, что МТ представляет собой сложное образование, различные отделы которого (передние, средние, задние) функционально неравнозначны и вносят специфический вклад в обеспечение межполушарного взаимодействия [19,20].Существуют и врожденные аномалии МТ. Агенезия мозолистого тела (АМТ) — полное отсутствие основной комиссурной спайки головного мозга. Гипоплазия -отсутствие только задней спайки и укорочение МТ. Помимо перечисленных выше пороков развития комиссурной системы мозга может наблюдаться также утолщение или истончение МТ. Такие клинические модели дают уникальную возможность для исследования межполушарных отношений и роли комиссурной системы [21-24].
С. 132]. После операции по «рассечению мозга» у больных не наблюдается отчетливых изменений темперамента, личности, общего интеллекта. Кроме того, описанный синдром является нестойким — со временем, в течение нескольких недель, выраженность симптомов существенно уменьшается, а иногда они вообще исчезают [1,18].Отечественные нейропсихологи исследовали больных с частичным рассечением МТ в результате удаления артериовенозных аневризм. Было установлено, что МТ представляет собой сложное образование, различные отделы которого (передние, средние, задние) функционально неравнозначны и вносят специфический вклад в обеспечение межполушарного взаимодействия [19,20].Существуют и врожденные аномалии МТ. Агенезия мозолистого тела (АМТ) — полное отсутствие основной комиссурной спайки головного мозга. Гипоплазия -отсутствие только задней спайки и укорочение МТ. Помимо перечисленных выше пороков развития комиссурной системы мозга может наблюдаться также утолщение или истончение МТ. Такие клинические модели дают уникальную возможность для исследования межполушарных отношений и роли комиссурной системы [21-24].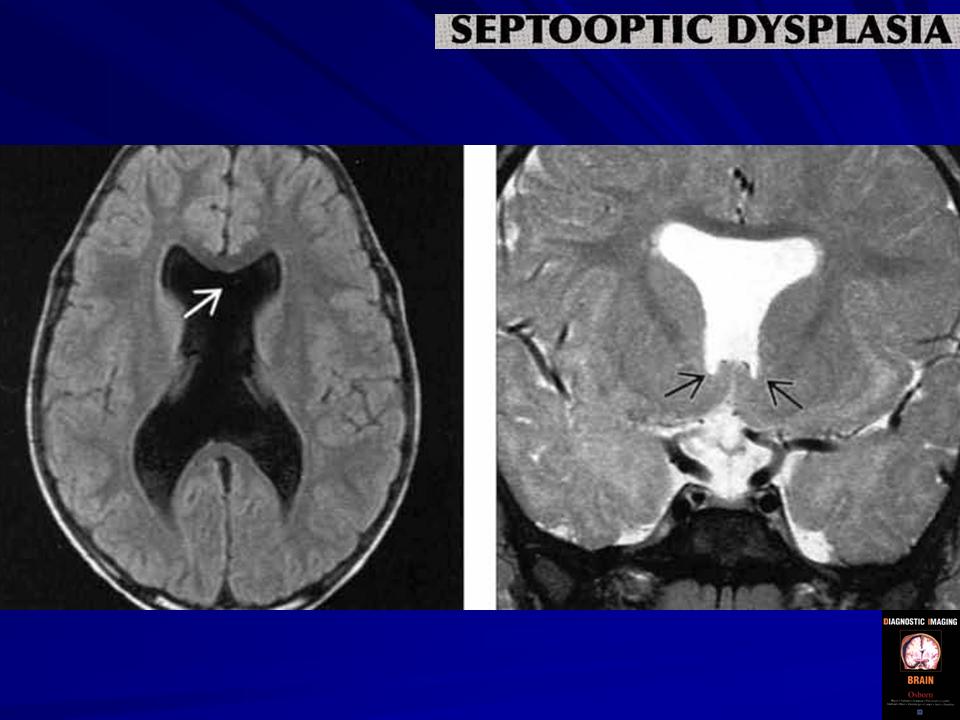 Аномальность мозолистого тела или дефицит в его функционировании может играть существенную роль в возникновении отклонений развития, неврологических и психических расстройств — аутизма, шизофрении, синдрома дефицита внимания и гиперактивности, дислексии и др. [25-31]. Анатомо-физиологические исследования детей с ранней гидроцефалией показывают, что расширение желудочков в этих случаях влечет атрофию мозговой ткани, особенно в области МТ [32]. Связь аномалий МТ с шизофренией в последнее время широко обсуждается в литературе [33]. При раннем начале шизофрении отмечается истончение МТ, причем истончаются в основном передняя и средняя его части. Утолщение МТ, наоборот, характерно для позднего начала заболевания и сочетается с хорошим про-193гнозом [24]. Прямой связи АМТ с психозами нет, но, возможно, МТ участвует в регуляции активности обоих полушарий, а недостаточность регуляции является одним из пусковых механизмов психотических расстройств [14]. Развитие компенсаторных связей при АМТ иногда идет неправильно, что также может сказываться на формировании симптомов психоза.
Аномальность мозолистого тела или дефицит в его функционировании может играть существенную роль в возникновении отклонений развития, неврологических и психических расстройств — аутизма, шизофрении, синдрома дефицита внимания и гиперактивности, дислексии и др. [25-31]. Анатомо-физиологические исследования детей с ранней гидроцефалией показывают, что расширение желудочков в этих случаях влечет атрофию мозговой ткани, особенно в области МТ [32]. Связь аномалий МТ с шизофренией в последнее время широко обсуждается в литературе [33]. При раннем начале шизофрении отмечается истончение МТ, причем истончаются в основном передняя и средняя его части. Утолщение МТ, наоборот, характерно для позднего начала заболевания и сочетается с хорошим про-193гнозом [24]. Прямой связи АМТ с психозами нет, но, возможно, МТ участвует в регуляции активности обоих полушарий, а недостаточность регуляции является одним из пусковых механизмов психотических расстройств [14]. Развитие компенсаторных связей при АМТ иногда идет неправильно, что также может сказываться на формировании симптомов психоза. Уменьшение рострума и ростральной части корпуса МТ описано при синдромах дефицита внимания и гиперактивности у детей [34]. В большинстве (71%) случаев гипоплазии МТ выявлена умственная отсталость [8].У детей с АМТ в возрасте до года нередко отмечаются судороги, отставание в моторном развитии, малая модуляция крика, нарушения сенсорных реакций, низкая коммуникабельность. У старших детей отмечают нарушения терморегуляции (гипотермию), дефицит координации, зрительной и слуховой памяти [35].Заметим, что исследования психических нарушений при АМТ имеют некоторые ограничения. Во-первых, часто это описания отдельных случаев. Во-вторых, результаты исследований детей с АМТ не всегда сопоставляются с результатами исследований контрольных пациентов с другими неврологическими нарушениями. Однако те исследования, которые включали контрольную группу, не обнаруживали у ее испытуемых дефицит, характерный для больных с АМТ [32, 36]. Осложняющим фактором во многих исследованиях является и то, что АМТ часто сопровождается различными неврологическими нарушениями, которые влияют на характер психического функционирования.
Уменьшение рострума и ростральной части корпуса МТ описано при синдромах дефицита внимания и гиперактивности у детей [34]. В большинстве (71%) случаев гипоплазии МТ выявлена умственная отсталость [8].У детей с АМТ в возрасте до года нередко отмечаются судороги, отставание в моторном развитии, малая модуляция крика, нарушения сенсорных реакций, низкая коммуникабельность. У старших детей отмечают нарушения терморегуляции (гипотермию), дефицит координации, зрительной и слуховой памяти [35].Заметим, что исследования психических нарушений при АМТ имеют некоторые ограничения. Во-первых, часто это описания отдельных случаев. Во-вторых, результаты исследований детей с АМТ не всегда сопоставляются с результатами исследований контрольных пациентов с другими неврологическими нарушениями. Однако те исследования, которые включали контрольную группу, не обнаруживали у ее испытуемых дефицит, характерный для больных с АМТ [32, 36]. Осложняющим фактором во многих исследованиях является и то, что АМТ часто сопровождается различными неврологическими нарушениями, которые влияют на характер психического функционирования. Существует более 50 расстройств, ассоциированных с АМТ [37].Остановимся подробнее на анализе особенностей психического функционирования при АМТ. Широко распространено мнение, что взрослые с АМТ не демонстрируют грубых нарушений межполушарного переноса сенсомоторной информации, которые характерны для пациентов с комиссуротомией [38, 39].Результаты исследования детей оказываются иными. Так, у детей 4 и 8 лет с полной АМТ была обнаружена неспособность называть объекты, ощупываемые субдоминантной рукой [40, 41]. В других исследованиях 8-летний ребенок с нормальным интеллектом демонстрировал дефицит межполушарного переноса тактильной информации при использовании доминантной руки [42].Для выявления нарушений взаимодействия полушарий при АМТ необходимы особым образом организованные эксперименты. Именно в таких условиях пациенты с АМТ демонстрируют дефицит межполушарного взаимодействия. У них отмечают затруднения и дезавтоматизацию при выполнении бимануальных движений, дефицит в межмануальной передаче пространственной информации (при тактильном прохождении лабиринта, в тактильно-конструктивных задачах) [43-45].
Существует более 50 расстройств, ассоциированных с АМТ [37].Остановимся подробнее на анализе особенностей психического функционирования при АМТ. Широко распространено мнение, что взрослые с АМТ не демонстрируют грубых нарушений межполушарного переноса сенсомоторной информации, которые характерны для пациентов с комиссуротомией [38, 39].Результаты исследования детей оказываются иными. Так, у детей 4 и 8 лет с полной АМТ была обнаружена неспособность называть объекты, ощупываемые субдоминантной рукой [40, 41]. В других исследованиях 8-летний ребенок с нормальным интеллектом демонстрировал дефицит межполушарного переноса тактильной информации при использовании доминантной руки [42].Для выявления нарушений взаимодействия полушарий при АМТ необходимы особым образом организованные эксперименты. Именно в таких условиях пациенты с АМТ демонстрируют дефицит межполушарного взаимодействия. У них отмечают затруднения и дезавтоматизацию при выполнении бимануальных движений, дефицит в межмануальной передаче пространственной информации (при тактильном прохождении лабиринта, в тактильно-конструктивных задачах) [43-45].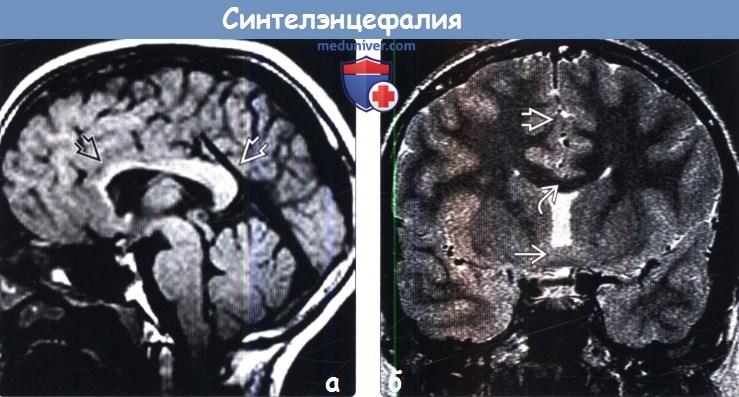 Систематический подход к тестированию испытуемых позволил проанализировать интеграцию полушар-ных и межполушарных функций. Дети с АМТ не отличались от нормальных детей и детей с другими неврологическими расстройствами по успешности выполнения мономануальных соматосенсорных тестов, требовавших вербального ответа на тактильные стимулы,однако им требовалось значительно больше времени для ответа при использовании субдоминантной руки. Напротив, в тесте на идентификацию фигур, требующем анализа перцептивных качеств предметов, дети с АМТ затрачивали значительно больше времени при использовании доминантной руки, чем испытуемые с неврологическими нарушениями [46]. Более длительное время выполнения некоторых психологических тестов испытуемыми с АМТ является следствием действия компенсаторных механизмов, т.е. функционирования альтернативных путей межполушарного переноса. При бимануальном восприятии текстуры и распознавании высоких пространственных частот дети с АМТ, в отличие от контрольных испытуемых, испытывали большие трудности.
Систематический подход к тестированию испытуемых позволил проанализировать интеграцию полушар-ных и межполушарных функций. Дети с АМТ не отличались от нормальных детей и детей с другими неврологическими расстройствами по успешности выполнения мономануальных соматосенсорных тестов, требовавших вербального ответа на тактильные стимулы,однако им требовалось значительно больше времени для ответа при использовании субдоминантной руки. Напротив, в тесте на идентификацию фигур, требующем анализа перцептивных качеств предметов, дети с АМТ затрачивали значительно больше времени при использовании доминантной руки, чем испытуемые с неврологическими нарушениями [46]. Более длительное время выполнения некоторых психологических тестов испытуемыми с АМТ является следствием действия компенсаторных механизмов, т.е. функционирования альтернативных путей межполушарного переноса. При бимануальном восприятии текстуры и распознавании высоких пространственных частот дети с АМТ, в отличие от контрольных испытуемых, испытывали большие трудности. Несмотря на данные о дефиците переноса тактильной информации у пациентов без передней части мозолистого тела, точность выполнения подобных заданий детьми с АМТ незначительно отличается от результатов детей с частичным отсутствием мозолистого тела. Детям с АМТ требовалось больше времени для ответа во всех бимануальных тестах на тактильное узнавание. Оценка текстуры оказалась единственным навыком, который у пациентов с АМТ был недоступен компенсации. Это говорит об определенной ограниченности компенсаторных возможностей при отсутствии мозолистого тела [46].Для передачи зрительной информации при АМТ наиболее подходящим кандидатом является передняя комиссура. Известно, что она играет важную роль в межполушарной передаче зрительной информации при распознавании фигур у обезьян [47, 48]. Передняя комиссура играет значительную роль в обеспечении функции зрения и у человека. При АМТ она может быть гораздо меньше по размеру или вообще отсутствовать, а может и увеличиваться [49]. Было проведено сравнение результатов обследования мальчика с АМТ и без передней комиссуры с результатами обследования мальчика без МТ, но с передней комиссурой.
Несмотря на данные о дефиците переноса тактильной информации у пациентов без передней части мозолистого тела, точность выполнения подобных заданий детьми с АМТ незначительно отличается от результатов детей с частичным отсутствием мозолистого тела. Детям с АМТ требовалось больше времени для ответа во всех бимануальных тестах на тактильное узнавание. Оценка текстуры оказалась единственным навыком, который у пациентов с АМТ был недоступен компенсации. Это говорит об определенной ограниченности компенсаторных возможностей при отсутствии мозолистого тела [46].Для передачи зрительной информации при АМТ наиболее подходящим кандидатом является передняя комиссура. Известно, что она играет важную роль в межполушарной передаче зрительной информации при распознавании фигур у обезьян [47, 48]. Передняя комиссура играет значительную роль в обеспечении функции зрения и у человека. При АМТ она может быть гораздо меньше по размеру или вообще отсутствовать, а может и увеличиваться [49]. Было проведено сравнение результатов обследования мальчика с АМТ и без передней комиссуры с результатами обследования мальчика без МТ, но с передней комиссурой. Оно показало, что у первого ребенка был затруднен перенос зрительной информации в левое полушарие, а тактильной — в правое, чего не наблюдалось во втором случае, что свидетельствует о возможности компенсации некоторых функций за счет передней комиссуры [44]. Эта комиссура может служить для межполушарной передачи достаточно сложной зрительной информации, обрабатываемой на корковом уровне (например, о форме букв и цифр). Пациенты с АМТ могли точно идентифицировать слова и абстрактные фигуры, предъявляемые в одно из двух полуполей зрения, но показывали явное преимущество левого полуполя при локализации стимулов внутри пространственной структуры. По-видимому, у индивидов с АМТ только информация о форме фигуры передается через переднюю комиссуру, а информация о положении — нет [50].Способность интегрировать зрительную информацию была исследована на 3 испытуемых с АМТ [24]. Испытуемыми были 39-летняя мать и две ее дочери, 11 и 12 лет. Им предлагалось называть и сравнивать цвета, цифры от 0 до 9 и буквы, предъявляемые попеременно в левое/правое зрительные полуполя или билатерально, а также называть слова из 6 букв (например, BARROW, SEALED, COTTON), половины которых предъявлялись194также попеременно в левое/правое зрительное полупо-ля или билатерально.
Оно показало, что у первого ребенка был затруднен перенос зрительной информации в левое полушарие, а тактильной — в правое, чего не наблюдалось во втором случае, что свидетельствует о возможности компенсации некоторых функций за счет передней комиссуры [44]. Эта комиссура может служить для межполушарной передачи достаточно сложной зрительной информации, обрабатываемой на корковом уровне (например, о форме букв и цифр). Пациенты с АМТ могли точно идентифицировать слова и абстрактные фигуры, предъявляемые в одно из двух полуполей зрения, но показывали явное преимущество левого полуполя при локализации стимулов внутри пространственной структуры. По-видимому, у индивидов с АМТ только информация о форме фигуры передается через переднюю комиссуру, а информация о положении — нет [50].Способность интегрировать зрительную информацию была исследована на 3 испытуемых с АМТ [24]. Испытуемыми были 39-летняя мать и две ее дочери, 11 и 12 лет. Им предлагалось называть и сравнивать цвета, цифры от 0 до 9 и буквы, предъявляемые попеременно в левое/правое зрительные полуполя или билатерально, а также называть слова из 6 букв (например, BARROW, SEALED, COTTON), половины которых предъявлялись194также попеременно в левое/правое зрительное полупо-ля или билатерально..jpg) Выявилось, что все испытуемые способны называть цвета, буквы и цифры, предъявляемые в оба зрительных полуполя, а также способны определить, похожи или отличаются буквы и цифры, предъявляемые билатерально. Но у них наблюдались большие трудности при сравнении предъявляемых билатерально цветов. При этом сравнение было значительно успешнее в случаях, когда цветовые стимулы находились ближе к средней линии, чем при их большем удалении от центра. Возможно, при АМТ информация о цвете не передается в противоположное полушарие вообще или передается в искаженном виде. При распознавании разделенных по центру 6-буквенных слов испытуемые были склонны воспринимать их целиком. Эти данные отличаются от результатов выполнения аналогичного задания пациентами с комиссуро-томией, которые имеют тенденцию называть такие слова как пару самостоятельных слов [51].Дети с АМТ могут осуществлять межполушарное сравнение одиночных букв, но обнаруживают значительный дефицит при сравнении пространственных паттернов, состоящих из 5 точек и предъявляемых поочередно в правое и левое зрительные полуполя.
Выявилось, что все испытуемые способны называть цвета, буквы и цифры, предъявляемые в оба зрительных полуполя, а также способны определить, похожи или отличаются буквы и цифры, предъявляемые билатерально. Но у них наблюдались большие трудности при сравнении предъявляемых билатерально цветов. При этом сравнение было значительно успешнее в случаях, когда цветовые стимулы находились ближе к средней линии, чем при их большем удалении от центра. Возможно, при АМТ информация о цвете не передается в противоположное полушарие вообще или передается в искаженном виде. При распознавании разделенных по центру 6-буквенных слов испытуемые были склонны воспринимать их целиком. Эти данные отличаются от результатов выполнения аналогичного задания пациентами с комиссуро-томией, которые имеют тенденцию называть такие слова как пару самостоятельных слов [51].Дети с АМТ могут осуществлять межполушарное сравнение одиночных букв, но обнаруживают значительный дефицит при сравнении пространственных паттернов, состоящих из 5 точек и предъявляемых поочередно в правое и левое зрительные полуполя. Эти данные свидетельствуют о том, что при отсутствии МТ лишь ограниченное количество простых зрительных образов может быть передано в другое полушарие. Эта передача осуществляется, по-видимому, через переднюю комиссуру и значительно ограничена [52].Существуют и данные о том, что МТ участвует в обеспечении Струп-эффекта [53]. Струп-эффект наблюдался как у испытуемых с АМТ, так и в контрольной группе здоровых испытуемых, но статистически значимых различий между группами выявлено не было [53]. Вероятно, для обеспечения этого феномена МТ не является необходимым, и можно обойтись другими внекаллозальными путями.При исследовании способности лиц с АМТ к локализации звука в пространстве было выявлено ее снижение по сравнению с нормой, особенно заметное в тех случаях, когда звук подавался по средней линии. Кроме того, вербальные ответы в этих экспериментах были менее точными, чем мануальные [54]. Другие исследователи изучали мануальную реакцию на световые вспышки в условиях перекрестных и прямых ответов [55].
Эти данные свидетельствуют о том, что при отсутствии МТ лишь ограниченное количество простых зрительных образов может быть передано в другое полушарие. Эта передача осуществляется, по-видимому, через переднюю комиссуру и значительно ограничена [52].Существуют и данные о том, что МТ участвует в обеспечении Струп-эффекта [53]. Струп-эффект наблюдался как у испытуемых с АМТ, так и в контрольной группе здоровых испытуемых, но статистически значимых различий между группами выявлено не было [53]. Вероятно, для обеспечения этого феномена МТ не является необходимым, и можно обойтись другими внекаллозальными путями.При исследовании способности лиц с АМТ к локализации звука в пространстве было выявлено ее снижение по сравнению с нормой, особенно заметное в тех случаях, когда звук подавался по средней линии. Кроме того, вербальные ответы в этих экспериментах были менее точными, чем мануальные [54]. Другие исследователи изучали мануальную реакцию на световые вспышки в условиях перекрестных и прямых ответов [55].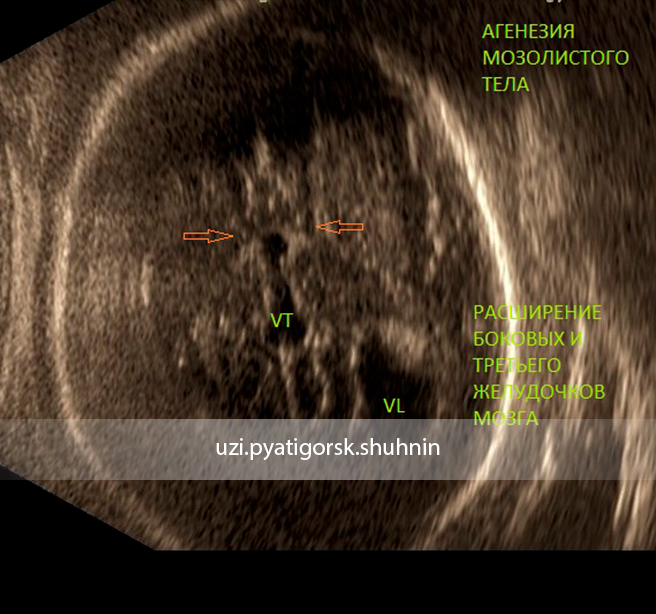 При этом они регистрировали электромиограмму с дистальных, проксимальных и аксиальных мышц. Эти исследования еще раз подтвердили, что различия в перекрестной и прямой реакциях у лиц с АМТ больше, чем в норме. Было также обнаружено, что МТ вносит свой вклад в выполнение быстрых контралатеральных дистальных и унилатеральных проксимальных ответов. Другие реакции, видимо, координируются через нижележащие пути.МТ участвует в бимануальной координации движений. Такая координация требует быстрого обмена информацией между полушариями головного мозга. МТ может участвовать в передаче моторных команд и эфферентной информации из одного полушария в другое, в реализации сенсорной обратной связи [12].При перерезке передней части МТ нарушалось выполнение заданий, требующих согласованных движе-ний обеих рук для рисования линий, варьирующих по степени наклона. В норме испытуемые были способны рисовать плавные линии в пробах, требующих как одинаковых, так и различных скоростей движений рук; при отсутствии визуальной обратной связи выполнение изменялось незначительно.
При этом они регистрировали электромиограмму с дистальных, проксимальных и аксиальных мышц. Эти исследования еще раз подтвердили, что различия в перекрестной и прямой реакциях у лиц с АМТ больше, чем в норме. Было также обнаружено, что МТ вносит свой вклад в выполнение быстрых контралатеральных дистальных и унилатеральных проксимальных ответов. Другие реакции, видимо, координируются через нижележащие пути.МТ участвует в бимануальной координации движений. Такая координация требует быстрого обмена информацией между полушариями головного мозга. МТ может участвовать в передаче моторных команд и эфферентной информации из одного полушария в другое, в реализации сенсорной обратной связи [12].При перерезке передней части МТ нарушалось выполнение заданий, требующих согласованных движе-ний обеих рук для рисования линий, варьирующих по степени наклона. В норме испытуемые были способны рисовать плавные линии в пробах, требующих как одинаковых, так и различных скоростей движений рук; при отсутствии визуальной обратной связи выполнение изменялось незначительно.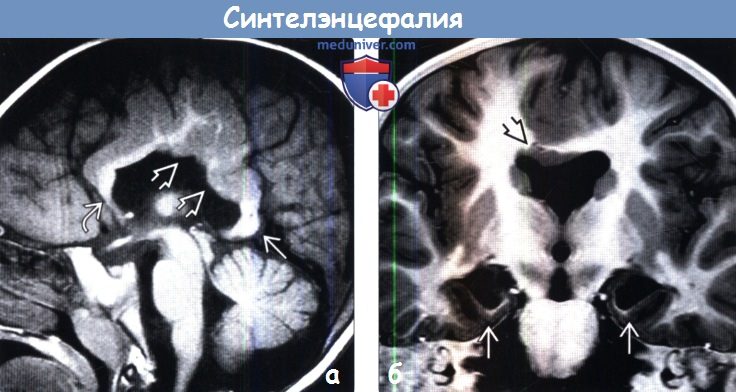 А испытуемые с частичной комиссуротомией сталкивались со значительными затруднениями при рисовании линий в пробах, требующих асимметричных движений рук, и их выполнение значительно ухудшалось, когда визуальная обратная связь была недоступна [12]. Приобретение таких моторных навыков у детей связано с миелинизацией МТ [56]. Испытуемые 10 и 20 лет выполняли аналогичные задания без затруднений, в то время как 6-летние дети демонстрировали результаты, сходные с результатами взрослых, перенесших комиссуротомию. Маленькие дети работали медленнее и делали больше ошибок, чем старшие дети, в основном в тех случаях, когда были необходимы различные скорости рук при рисовании наклонных линий. Дефицит бимануальной координации, возможно, был связан с неэффективной передачей моторной информации между полушариями при неполной миелинизации МТ [56].Нарушения выполнения бимануальных проб также были констатированы у пациентов с расстройствами чтения [57]. Дети и взрослые с дислексией выполняли задания так же, как пациенты с комиссуротомией и более младшие дети без дислексии [58].
А испытуемые с частичной комиссуротомией сталкивались со значительными затруднениями при рисовании линий в пробах, требующих асимметричных движений рук, и их выполнение значительно ухудшалось, когда визуальная обратная связь была недоступна [12]. Приобретение таких моторных навыков у детей связано с миелинизацией МТ [56]. Испытуемые 10 и 20 лет выполняли аналогичные задания без затруднений, в то время как 6-летние дети демонстрировали результаты, сходные с результатами взрослых, перенесших комиссуротомию. Маленькие дети работали медленнее и делали больше ошибок, чем старшие дети, в основном в тех случаях, когда были необходимы различные скорости рук при рисовании наклонных линий. Дефицит бимануальной координации, возможно, был связан с неэффективной передачей моторной информации между полушариями при неполной миелинизации МТ [56].Нарушения выполнения бимануальных проб также были констатированы у пациентов с расстройствами чтения [57]. Дети и взрослые с дислексией выполняли задания так же, как пациенты с комиссуротомией и более младшие дети без дислексии [58]. У лиц с врожденной или приобретенной аномалией МТ изменяется временная организация выполнения различных бимануальных программ [59]. Пациентам с АМТ и с комиссуротомией предлагалось выполнить следующие задания: 1) открывание выдвижного ящика, при котором одна рука выдвигает ящик, в то время как другая берет из него маленький предмет; 2) ритмические вращательные движения рук, которые выполняются параллельно или в противоположных направлениях. Пациенты с АМТ в основном оптимально выполняли первое задание, но демонстрировали сильную тенденцию к десинхронизации вращательных движений, особенно совершаемых в противоположных направлениях. Однако такие проблемы испытывали лишь некоторые пациенты. По-видимому, при АМТ для регулирования временной синхронизации в бимануальных движениях могут быть использованы компенсаторные механизмы [59]. При изолированном поражении МТ может наблюдаться апраксия в левых конечностях [60, 61].Ухудшение качества межполушарной интеграции комплексной информации может влиять на некоторые аспекты развития интеллекта и сложной когнитивной деятельности.
У лиц с врожденной или приобретенной аномалией МТ изменяется временная организация выполнения различных бимануальных программ [59]. Пациентам с АМТ и с комиссуротомией предлагалось выполнить следующие задания: 1) открывание выдвижного ящика, при котором одна рука выдвигает ящик, в то время как другая берет из него маленький предмет; 2) ритмические вращательные движения рук, которые выполняются параллельно или в противоположных направлениях. Пациенты с АМТ в основном оптимально выполняли первое задание, но демонстрировали сильную тенденцию к десинхронизации вращательных движений, особенно совершаемых в противоположных направлениях. Однако такие проблемы испытывали лишь некоторые пациенты. По-видимому, при АМТ для регулирования временной синхронизации в бимануальных движениях могут быть использованы компенсаторные механизмы [59]. При изолированном поражении МТ может наблюдаться апраксия в левых конечностях [60, 61].Ухудшение качества межполушарной интеграции комплексной информации может влиять на некоторые аспекты развития интеллекта и сложной когнитивной деятельности. Вместе с тем сохранные межполушар-ные связи часто обеспечивают реорганизацию функций, необходимую в случае унилатерального мозгового повреждения. Различные исследователи указывают на большую вариабельность умственных способностей детей с АМТ, хотя большинство из них имеет нормальный уровень интеллекта. У некоторых детей с изолированной АМТ (частичной или полной) нормальный уровень интеллекта в дошкольном возрасте мог иметь тенденцию понижаться в школьном возрасте, а также появлялись вялость и неустойчивость внимания. Стати-195стически значимых различий между когнитивным развитием детей с полной и частичной АМТ выявлено не было. У испытуемых с АМТ в тестах на категории выявлялись затруднения в формировании концепции решения. Значительные трудности были заметны при сравнении геометрических форм и цветов, особенно после успешного выполнения заданий, для решения которых была необходима опора на количество или цифры [62].Таким образом, различные работы свидетельствуют о том, что когнитивные трудности при АМТ не являются универсальными; дефицит возникает скорее при выполнении новых и сложных когнитивных задач.
Вместе с тем сохранные межполушар-ные связи часто обеспечивают реорганизацию функций, необходимую в случае унилатерального мозгового повреждения. Различные исследователи указывают на большую вариабельность умственных способностей детей с АМТ, хотя большинство из них имеет нормальный уровень интеллекта. У некоторых детей с изолированной АМТ (частичной или полной) нормальный уровень интеллекта в дошкольном возрасте мог иметь тенденцию понижаться в школьном возрасте, а также появлялись вялость и неустойчивость внимания. Стати-195стически значимых различий между когнитивным развитием детей с полной и частичной АМТ выявлено не было. У испытуемых с АМТ в тестах на категории выявлялись затруднения в формировании концепции решения. Значительные трудности были заметны при сравнении геометрических форм и цветов, особенно после успешного выполнения заданий, для решения которых была необходима опора на количество или цифры [62].Таким образом, различные работы свидетельствуют о том, что когнитивные трудности при АМТ не являются универсальными; дефицит возникает скорее при выполнении новых и сложных когнитивных задач. Исследования речевых функций у детей с АМТ (при нормальном интеллекте) выявляют у них специфическое ухудшение фонологических процессов (неспособность формулировать и узнавать рифмованный стих) [63]. Иногда обнаруживается специфический дефицит в понимании синтаксической и металингвистической организации речи [64]. Есть данные о случаях задержек речевого развития у детей с АМТ, а также о появляющихся с возрастом трудностях аргументации [62]. Таким образом, хотя нормальный интеллект детей с АМТ может обеспечивать адекватное понимание лексической организации текста, они в некоторых случаях обнаруживают своеобразный языковый дефицит. Он отмечается, если дети с АМТ встречаются с комплексом новых слов, нуждающихся в фонологическом объяснении; с заданиями, характеризующимися сложным синтаксисом или при необходимости понимания переносного смысла пословиц и метафор.При АМТ изменяется и восприятие просодических компонентов речи [38]. Это демонстрируют следующие эксперименты. В первом задании испытуемым предъявлялись аудиозаписи 16 семантически нейтральных простых предложений, сделанные женским голосом с одной из 4 эмоционально-насыщенных интонаций (счастье, грусть, злость, удивление).
Исследования речевых функций у детей с АМТ (при нормальном интеллекте) выявляют у них специфическое ухудшение фонологических процессов (неспособность формулировать и узнавать рифмованный стих) [63]. Иногда обнаруживается специфический дефицит в понимании синтаксической и металингвистической организации речи [64]. Есть данные о случаях задержек речевого развития у детей с АМТ, а также о появляющихся с возрастом трудностях аргументации [62]. Таким образом, хотя нормальный интеллект детей с АМТ может обеспечивать адекватное понимание лексической организации текста, они в некоторых случаях обнаруживают своеобразный языковый дефицит. Он отмечается, если дети с АМТ встречаются с комплексом новых слов, нуждающихся в фонологическом объяснении; с заданиями, характеризующимися сложным синтаксисом или при необходимости понимания переносного смысла пословиц и метафор.При АМТ изменяется и восприятие просодических компонентов речи [38]. Это демонстрируют следующие эксперименты. В первом задании испытуемым предъявлялись аудиозаписи 16 семантически нейтральных простых предложений, сделанные женским голосом с одной из 4 эмоционально-насыщенных интонаций (счастье, грусть, злость, удивление). Испытуемые должны были соотносить предложения с одной из четырех картинок, на которых были нарисованы эмоционально-экспрессивные лица. Второе задание содержало 20 обычных предложений и 20 выражений метафорического содержания, которые были одинаковы по длине и грамматической структуре. Испытуемый должен был выбрать одну из 4 картинок, наиболее соответствующую данному выражению. Третье задание включало в себя толкование пословиц, причем сначала испытуемый должен был написать свою версию ответа, а потом ему предлагалось выбрать один из четырех ответов, предложенных экспериментатором. Оказалось, что испытуемые с АМТ хуже, чем контрольная группа, идентифицировали эмоционально-просодические компоненты речи. У них наблюдалось явное снижение точности идентификации метафоричных выражений. При интерпретации пословиц испытуемые с АМТ были менее точны в структурном и синтаксическом оформлении высказываний. Вероятно, испытуемые с АМТ испытывают значительные трудности именно в тех аспектах речевых процессов, которые наиболее важны для социальной коммуникации [38].
Испытуемые должны были соотносить предложения с одной из четырех картинок, на которых были нарисованы эмоционально-экспрессивные лица. Второе задание содержало 20 обычных предложений и 20 выражений метафорического содержания, которые были одинаковы по длине и грамматической структуре. Испытуемый должен был выбрать одну из 4 картинок, наиболее соответствующую данному выражению. Третье задание включало в себя толкование пословиц, причем сначала испытуемый должен был написать свою версию ответа, а потом ему предлагалось выбрать один из четырех ответов, предложенных экспериментатором. Оказалось, что испытуемые с АМТ хуже, чем контрольная группа, идентифицировали эмоционально-просодические компоненты речи. У них наблюдалось явное снижение точности идентификации метафоричных выражений. При интерпретации пословиц испытуемые с АМТ были менее точны в структурном и синтаксическом оформлении высказываний. Вероятно, испытуемые с АМТ испытывают значительные трудности именно в тех аспектах речевых процессов, которые наиболее важны для социальной коммуникации [38]. Есть также данные о том, что пациентам с АМТ с нормальной экспрессивной речью были свойственны такие особенности, какведение бессмысленных или не касающихся темы разговоров [65].Для лиц с АМТ характерен и некоторый эмоциональный дефицит. При исследовании детей с частичной АМТ, полной АМТ обнаружился недостаток эмоциональной коммуникации, который был общим для всех трех групп [66]. Хотя типичные для детей с аутизмом симптомы (социальное равнодушие, дисфоричное настроение и раздражительность) в целом не характерны для испытуемых с полной и частичной АМТ, однако родители детей с АМТ часто сообщают о том, что обычно трудно узнать, что их дети чувствуют в этот момент или эмоционально переживают [66]. Некоторые авторы считают, что это может быть связано с алекситимией, общими характеристиками которой являются недостаток эмоциональной экспрессии, бедность фантазии, повышенная зависимость чувств от внешних событий. Связь алекситимии и межполушар-ного дефицита была показана и при исследовании двенадцати пациентов с комиссуротомией [67].
Есть также данные о том, что пациентам с АМТ с нормальной экспрессивной речью были свойственны такие особенности, какведение бессмысленных или не касающихся темы разговоров [65].Для лиц с АМТ характерен и некоторый эмоциональный дефицит. При исследовании детей с частичной АМТ, полной АМТ обнаружился недостаток эмоциональной коммуникации, который был общим для всех трех групп [66]. Хотя типичные для детей с аутизмом симптомы (социальное равнодушие, дисфоричное настроение и раздражительность) в целом не характерны для испытуемых с полной и частичной АМТ, однако родители детей с АМТ часто сообщают о том, что обычно трудно узнать, что их дети чувствуют в этот момент или эмоционально переживают [66]. Некоторые авторы считают, что это может быть связано с алекситимией, общими характеристиками которой являются недостаток эмоциональной экспрессии, бедность фантазии, повышенная зависимость чувств от внешних событий. Связь алекситимии и межполушар-ного дефицита была показана и при исследовании двенадцати пациентов с комиссуротомией [67].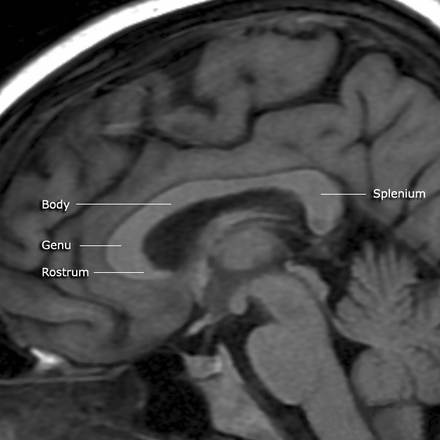 Исследователи предположили, что недостаток взаимосвязи между правым полушарием, отвечающим за аффективный опыт, и левым, контролирующим вербализацию, выражается в неспособности выражать эмоции. Уменьшение вербальной экспрессии эмоций у лиц с АМТ обнаруживается при нормальном уровне интеллекта [68]. Сходные результаты у пациентов с комиссуротомией и лиц с АМТ позволяют предположить, что мозолистое тело играет важную роль в мозговом обеспечении способности переживать и описывать свои эмоции.У детей с АМТ могут наблюдаться и некоторые психосоциальные трудности. Некоторые авторы отмечают, что родители говорят о бедной социальной интуиции таких детей, о дефиците межличностных отношений в семье [62]. Они сообщают, что их дети не понимают шуток, не всегда способны выразить свое мнение в новых социальных ситуациях.Исследования показывают, что именно дефицит ряда аспектов социальной перцепции является наиболее явным последствием отсутствия МТ. Особый интерес вызывает тот факт, что АМТ влечет за собой нарушение восприятия юмора и, следовательно, дефицит в коммуникативной сфере [69-73].
Исследователи предположили, что недостаток взаимосвязи между правым полушарием, отвечающим за аффективный опыт, и левым, контролирующим вербализацию, выражается в неспособности выражать эмоции. Уменьшение вербальной экспрессии эмоций у лиц с АМТ обнаруживается при нормальном уровне интеллекта [68]. Сходные результаты у пациентов с комиссуротомией и лиц с АМТ позволяют предположить, что мозолистое тело играет важную роль в мозговом обеспечении способности переживать и описывать свои эмоции.У детей с АМТ могут наблюдаться и некоторые психосоциальные трудности. Некоторые авторы отмечают, что родители говорят о бедной социальной интуиции таких детей, о дефиците межличностных отношений в семье [62]. Они сообщают, что их дети не понимают шуток, не всегда способны выразить свое мнение в новых социальных ситуациях.Исследования показывают, что именно дефицит ряда аспектов социальной перцепции является наиболее явным последствием отсутствия МТ. Особый интерес вызывает тот факт, что АМТ влечет за собой нарушение восприятия юмора и, следовательно, дефицит в коммуникативной сфере [69-73]. В одном из исследований для выявления уровня восприятия юмора у таких больных был использован тест Browneli и Bihrle [69, 70]. Юмористический тест состоял из невербальных и вербальных субтестов. Каждый невербальный субтест включал три последовательные комические картинки без подписей, к которым предлагалось два варианта концовок. Испытуемые должны были выбрать более смешной. Вербальный субтест состоял из шуточного предложения, к которому предлагалось пять вариантов окончаний — перед испытуемым также стояла задача выбрать наиболее подходящий вариант.В исследовании участвовали 16 испытуемых с АМТ в возрасте от 14 до 55 лет и 31 здоровый испытуемый такого же возраста и уровня умственного развития. Результаты вербальных субтестов выявили дефицит в понимании юмора у лиц с АМТ. Однако при выполнении субтестов на восприятие и понимание невербальных аспектов юмора сложностей у этих испытуемых обнаружено не было. Характерным также было то, что196испытуемые с АМТ придерживались первоначального решения и не изменяли его, даже когда допускали ошибки, в отличие от контрольной группы, демонстрирующей достаточную пластичность своих решений.
В одном из исследований для выявления уровня восприятия юмора у таких больных был использован тест Browneli и Bihrle [69, 70]. Юмористический тест состоял из невербальных и вербальных субтестов. Каждый невербальный субтест включал три последовательные комические картинки без подписей, к которым предлагалось два варианта концовок. Испытуемые должны были выбрать более смешной. Вербальный субтест состоял из шуточного предложения, к которому предлагалось пять вариантов окончаний — перед испытуемым также стояла задача выбрать наиболее подходящий вариант.В исследовании участвовали 16 испытуемых с АМТ в возрасте от 14 до 55 лет и 31 здоровый испытуемый такого же возраста и уровня умственного развития. Результаты вербальных субтестов выявили дефицит в понимании юмора у лиц с АМТ. Однако при выполнении субтестов на восприятие и понимание невербальных аспектов юмора сложностей у этих испытуемых обнаружено не было. Характерным также было то, что196испытуемые с АМТ придерживались первоначального решения и не изменяли его, даже когда допускали ошибки, в отличие от контрольной группы, демонстрирующей достаточную пластичность своих решений. Возможно, дефицит в понимании юмора при АМТ связан с нарушением выявления и конструирования подтекста на интерпретативном уровне, а также установления соответствия между частями предложения или текста. Эта гипотеза подтверждается и тем, что испытуемые не пытались использовать различные варианты, предложенные в субтестах, а руководствовались первым попавшимся. Нарушение коммуникативной функции при АМТ также проявляется в исследовании структуры рассказов ТАТ [74]. Оценивалось три параметра, по которым испытуемые строили свой рассказ: логика рассказа, его социальная значимость и содержание. Шесть испытуемых с полной или частичной АМТ продемонстрировали низкие показатели по всем трем параметрам. Эти результаты показывают, что индивиды с полной АМТ испытывают трудности в понимании рассказов, сцен социального характера и в составлении устных рассказов на социальные темы [74]. Таким образом, роль МТ в процессах, касающихся социального опыта, представляется очень важной.Обобщая приведенные выше данные, можно сказать, что нарушения межполушарного взаимодействия не ведут к формированию классического «синдрома расщепления».
Возможно, дефицит в понимании юмора при АМТ связан с нарушением выявления и конструирования подтекста на интерпретативном уровне, а также установления соответствия между частями предложения или текста. Эта гипотеза подтверждается и тем, что испытуемые не пытались использовать различные варианты, предложенные в субтестах, а руководствовались первым попавшимся. Нарушение коммуникативной функции при АМТ также проявляется в исследовании структуры рассказов ТАТ [74]. Оценивалось три параметра, по которым испытуемые строили свой рассказ: логика рассказа, его социальная значимость и содержание. Шесть испытуемых с полной или частичной АМТ продемонстрировали низкие показатели по всем трем параметрам. Эти результаты показывают, что индивиды с полной АМТ испытывают трудности в понимании рассказов, сцен социального характера и в составлении устных рассказов на социальные темы [74]. Таким образом, роль МТ в процессах, касающихся социального опыта, представляется очень важной.Обобщая приведенные выше данные, можно сказать, что нарушения межполушарного взаимодействия не ведут к формированию классического «синдрома расщепления». Пока описан только один нейропсихо-логический синдром нарушения межполушарного взаимодействия, связанный с повреждением МТ. Будут ли нейропсихологические синдромы при АМТ или других вариантах дефицитарности межполушарного взаимодействия отличаться от синдрома «расщепленного мозга»? Ответы на некоторые из этих вопросов были получены при проведении нейропсихологических исследований, однако другие пока остаются не до конца проясненными [75, 76]. Поэтому нашей первоочередной целью стало исследование особенностей высших психических функций при различных формах патологии МТ.Нами было обследовано 29 больных с различной патологией МТ в возрасте от 5 до 38 лет. 14 человек (дети) были в возрасте от 5 до 13 лет. Среди них у 11 была констатирована АМТ, у 3 — гипоплазия МТ. 15 человек (взрослые) были в возрасте от 18 до 38 лет. Среди них у 4 больных была полная АМТ, у 2 — частичная АМТ, у 1 — аплазия, у 1 — киста МТ, у 7 — арте-риовенозная мальформация МТ. У двух больных с АМТ из взрослой группы отмечались эпилептические приступы.
Пока описан только один нейропсихо-логический синдром нарушения межполушарного взаимодействия, связанный с повреждением МТ. Будут ли нейропсихологические синдромы при АМТ или других вариантах дефицитарности межполушарного взаимодействия отличаться от синдрома «расщепленного мозга»? Ответы на некоторые из этих вопросов были получены при проведении нейропсихологических исследований, однако другие пока остаются не до конца проясненными [75, 76]. Поэтому нашей первоочередной целью стало исследование особенностей высших психических функций при различных формах патологии МТ.Нами было обследовано 29 больных с различной патологией МТ в возрасте от 5 до 38 лет. 14 человек (дети) были в возрасте от 5 до 13 лет. Среди них у 11 была констатирована АМТ, у 3 — гипоплазия МТ. 15 человек (взрослые) были в возрасте от 18 до 38 лет. Среди них у 4 больных была полная АМТ, у 2 — частичная АМТ, у 1 — аплазия, у 1 — киста МТ, у 7 — арте-риовенозная мальформация МТ. У двух больных с АМТ из взрослой группы отмечались эпилептические приступы..jpg) Все обследованные больные были правору-кими. Во всех случаях проводилась строгая верификация патологии МТ с помощью МРТ и ангиографии.В неврологическом статусе обследованных не отмечалось отчетливых знаков очагового поражения ЦНС, однако выявлялась негрубая неврологическая симптоматика: мышечная гипотония, признаки гидроцефаль-ного синдрома в стадии субкомпенсации, отсутствие или снижение конвергенции.При комплексном клинико-нейропсихологическом обследовании у всех больных обнаруживались нарушения высших психических функций разной степени выраженности.Во-первых, отмечались нарушения зрительно-пространственных функций: структурно-топологические ошибки при копировании и воспроизведении по памяти сложных геометрических фигур типа фигуры Рея-Тейлора; левостороннее игнорирование или тенденция к нему; дисметрические ошибки (±5 мин, ±1 ч) при определении времени по «слепым» часам; фрагментарные ошибки при узнавании недорисованных предметов; трудности рисования объемных изображений (куба, домика).
Все обследованные больные были правору-кими. Во всех случаях проводилась строгая верификация патологии МТ с помощью МРТ и ангиографии.В неврологическом статусе обследованных не отмечалось отчетливых знаков очагового поражения ЦНС, однако выявлялась негрубая неврологическая симптоматика: мышечная гипотония, признаки гидроцефаль-ного синдрома в стадии субкомпенсации, отсутствие или снижение конвергенции.При комплексном клинико-нейропсихологическом обследовании у всех больных обнаруживались нарушения высших психических функций разной степени выраженности.Во-первых, отмечались нарушения зрительно-пространственных функций: структурно-топологические ошибки при копировании и воспроизведении по памяти сложных геометрических фигур типа фигуры Рея-Тейлора; левостороннее игнорирование или тенденция к нему; дисметрические ошибки (±5 мин, ±1 ч) при определении времени по «слепым» часам; фрагментарные ошибки при узнавании недорисованных предметов; трудности рисования объемных изображений (куба, домика). Подавляющее большинство выявленных ошибок или затруднений были аналогичны симптомам, наблюдаемым в клинике локальных поражений мозга при заинтересованности правого полушария. Интересно отметить, что не было выявлено никакой разницы при копировании и рисовании правой и левой руками.На втором месте по частоте встречаемости стояли негрубые, но отчетливые нарушения памяти по модально-неспецифическому типу. Они возникали преимущественно из-за повышенной тормозимости следов в условиях интерференции. В слухоречевой модальности встречались ошибки удержания порядка элементов стимульного ряда, а в зрительной модальности у некоторых испытуемых отмечалась реверсия исходного стимульного ряда (его «зеркальное» воспроизведение) или реверсии отдельных стимулов.В сфере праксиса у большинства испытуемых была констатирована сохранность выполнения реципрокной координации. Лишь несколько больных (в основном, детского возраста) демонстрировали отставание левой руки или ее игнорирование при выполнении этой пробы.
Подавляющее большинство выявленных ошибок или затруднений были аналогичны симптомам, наблюдаемым в клинике локальных поражений мозга при заинтересованности правого полушария. Интересно отметить, что не было выявлено никакой разницы при копировании и рисовании правой и левой руками.На втором месте по частоте встречаемости стояли негрубые, но отчетливые нарушения памяти по модально-неспецифическому типу. Они возникали преимущественно из-за повышенной тормозимости следов в условиях интерференции. В слухоречевой модальности встречались ошибки удержания порядка элементов стимульного ряда, а в зрительной модальности у некоторых испытуемых отмечалась реверсия исходного стимульного ряда (его «зеркальное» воспроизведение) или реверсии отдельных стимулов.В сфере праксиса у большинства испытуемых была констатирована сохранность выполнения реципрокной координации. Лишь несколько больных (в основном, детского возраста) демонстрировали отставание левой руки или ее игнорирование при выполнении этой пробы. При выполнении пробы на перенос позы пальцев по кинестетическому образцу (т.е. без зрительного контроля) у всех испытуемых был снижен темп деятельности; у некоторых испытуемых из детской группы данная проба вызывала отчетливые билатеральные затруднения. Интересно, что все дети и взрослые с АМТ жаловались на трудности обучения катанию на велосипеде, неуверенность в беге, частые падения.В пробах на тактильный гнозис (типа доски Сегена) испытуемый должен был без зрительного контроля ощупать фигурку одной рукой, а соответствующую ей ячейку найти другой рукой. В этом задании у некоторых испытуемых отмечались негрубые трудности называния (аналог аномии) ощупываемых фигурок и отыскивания ячеек левой рукой.Речь у всех больных с АМТ была сохранна, однако практически все они рассказывали о том, что научились говорить достаточно поздно. При чтении (особенно в детской группе) отмечалось интонационное игнорирование точек, неправильные ударения в словах. В письме часто встречались пропуски букв, особенно гласных.
При выполнении пробы на перенос позы пальцев по кинестетическому образцу (т.е. без зрительного контроля) у всех испытуемых был снижен темп деятельности; у некоторых испытуемых из детской группы данная проба вызывала отчетливые билатеральные затруднения. Интересно, что все дети и взрослые с АМТ жаловались на трудности обучения катанию на велосипеде, неуверенность в беге, частые падения.В пробах на тактильный гнозис (типа доски Сегена) испытуемый должен был без зрительного контроля ощупать фигурку одной рукой, а соответствующую ей ячейку найти другой рукой. В этом задании у некоторых испытуемых отмечались негрубые трудности называния (аналог аномии) ощупываемых фигурок и отыскивания ячеек левой рукой.Речь у всех больных с АМТ была сохранна, однако практически все они рассказывали о том, что научились говорить достаточно поздно. При чтении (особенно в детской группе) отмечалось интонационное игнорирование точек, неправильные ударения в словах. В письме часто встречались пропуски букв, особенно гласных..jpg) У одной взрослой больной с кистой МТ наблюдался своеобразный феномен насильственного «зеркального» письма ведущей рукой: независимо от желания больной рука при письме начинала двигаться в противоположную сторону.Интеллект у 27 испытуемых был полностью сохранен. Только у двух детей 9 и 10 лет, не демонстрировавших ранее никаких нарушений интеллекта, отмечались выраженные трудности при осмыслении и пересказе рассказов. При этом пересказ либо состоял из197отдельных, не связанных по смыслу фрагментов, либоконтроля. Следует подчеркнуть, что эти симптомы быпредставлял собой исходную сюжетную канву, напол-ли выражены негрубо и носили характер единичных ненную множественными конфабуляторными включе-проявлений. Многие из составляющих выявленного ниями. Интересно, что у одного из этих мальчиков од-синдрома носят правополушарный характер. Этот факт новременно с появлением подобных интеллектуальныхможет свидетельствовать о значительной функционарушений возникли симптомы правостороннего про-нальной роли правого полушария в обеспечении межстранственного игнорирования в рисуночных пробах.
У одной взрослой больной с кистой МТ наблюдался своеобразный феномен насильственного «зеркального» письма ведущей рукой: независимо от желания больной рука при письме начинала двигаться в противоположную сторону.Интеллект у 27 испытуемых был полностью сохранен. Только у двух детей 9 и 10 лет, не демонстрировавших ранее никаких нарушений интеллекта, отмечались выраженные трудности при осмыслении и пересказе рассказов. При этом пересказ либо состоял из197отдельных, не связанных по смыслу фрагментов, либоконтроля. Следует подчеркнуть, что эти симптомы быпредставлял собой исходную сюжетную канву, напол-ли выражены негрубо и носили характер единичных ненную множественными конфабуляторными включе-проявлений. Многие из составляющих выявленного ниями. Интересно, что у одного из этих мальчиков од-синдрома носят правополушарный характер. Этот факт новременно с появлением подобных интеллектуальныхможет свидетельствовать о значительной функционарушений возникли симптомы правостороннего про-нальной роли правого полушария в обеспечении межстранственного игнорирования в рисуночных пробах. полушарного взаимодействия. Можно предположить, Таким образом, у обследованных пациентов, осо-что именно правое полушарие запускает и контролирует бенно с АМТ, не выявлялось всего комплекса симпто-функционирование комиссурной системы мозга в онтогемов, составляющих классический синдром «расщеп-незе. С другой стороны, необходимо учитывать и разницу ленного мозга» или синдром частичной перерезки МТ.в функциональных связях комиссур с полушариями мозга Вместе с тем ряд черт демонстрировал определенное[77]. Можно ли говорить в этой связи о приоритетной сходство с этими клиническими моделями. Это, преждероли правой гемисферы? Каковы отличия между компенвсего, проявления унилатерального игнорирования,саторными и дефицитарными симптомами при патологии симптомы, сходные с аномией, трудности переноса позМТ? Ответы на эти непростые вопросы могут быть полуруки и выполнения тактильных проб без зрительногочены в дальнейших исследованиях.
полушарного взаимодействия. Можно предположить, Таким образом, у обследованных пациентов, осо-что именно правое полушарие запускает и контролирует бенно с АМТ, не выявлялось всего комплекса симпто-функционирование комиссурной системы мозга в онтогемов, составляющих классический синдром «расщеп-незе. С другой стороны, необходимо учитывать и разницу ленного мозга» или синдром частичной перерезки МТ.в функциональных связях комиссур с полушариями мозга Вместе с тем ряд черт демонстрировал определенное[77]. Можно ли говорить в этой связи о приоритетной сходство с этими клиническими моделями. Это, преждероли правой гемисферы? Каковы отличия между компенвсего, проявления унилатерального игнорирования,саторными и дефицитарными симптомами при патологии симптомы, сходные с аномией, трудности переноса позМТ? Ответы на эти непростые вопросы могут быть полуруки и выполнения тактильных проб без зрительногочены в дальнейших исследованиях.Ключевые слова
Авторы
Всего: 2Ссылки
Хомская Е.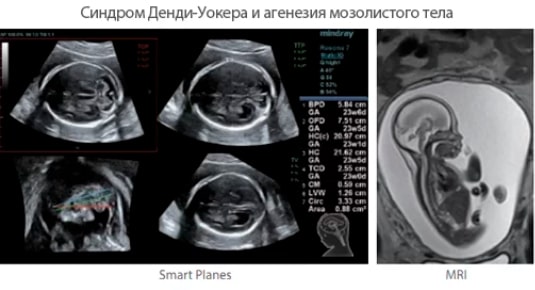 Д. Нейропсихология / Е.Д. Хомская. — М.: Психология, 2002. — 415 с.
Д. Нейропсихология / Е.Д. Хомская. — М.: Психология, 2002. — 415 с.
Yakovlev P. The myelogenetic cycles of regional maturation of the brain / P. Yakovlev, A. LeCours // Regional development of the Brain in early life / Ed. by A. Minkowski. — Oxford: Blackwell, 1967. — P. 3-69.
Barkovich A. Magnetic resonance imaging of normal and abnormal brain development / A. Barkovich, T. Maroldo // Topics in Magnetic Resonance Imaging. — 1993. — Vol. 5, № 2. — P. 96-122.
Giedd J. A quantitative MRI study of the CC in Children and adolescents / J. Giedd, J. Rumsey, F. Castellanos, J. Rajapakse, D. Kaysen, A. Vaituzis et al. // Developmental Brain Research. — 1996. — Vol. 91, № 2. — P. 274-280.
Thompson P. Growth patterns in the developing brain detected by using continuum mechanical tensor maps / P. Thompson, J.N. Giedd, R.P. Woods, D. MacDonald, A.C. Evans, A.W. Toga // Nature. — 2000. — Vol. 404, № 6774. — P. 190-193.
Pujol J. When dues humen brain development end? Evidence of CC growth up to adulthood / J. Pujol, P. Vendrell, С Jungue, J. Mart’-Vilalta, A. Capdevila // Annals of Neurology. — 1993. — Vol. 34, № 1. — P. 71-75.
When dues humen brain development end? Evidence of CC growth up to adulthood / J. Pujol, P. Vendrell, С Jungue, J. Mart’-Vilalta, A. Capdevila // Annals of Neurology. — 1993. — Vol. 34, № 1. — P. 71-75.
Geers M. Development of the humen CC during childhood and adolescence: A longitudinal MRI study / M. Geers, R. de Borst, T. Peijs, J. Giedd, J. Blumenthal, N. Jeffries, J. Rajapakse, A. Vaituzes, H. Liu // Progress in Neuro-psychopharmocology and Biological Psychiatry. — 1999. — Vol. 23, № 4.-P. 571-588.
Lamantia A. Cytological and quantitative characteristics of four cerebral commissures in the rhesus monkey / A. Lamantia, P. Rakic // J. of comparative neurology. — 1990. — Vol. 291, № 4. — P. 520-537.
Trevarthen C.B. Two mechanisms of vision in primates / C.B. Trevarthen // Psychol. Forsch. — 1968. — Vol. 31, № 2. — P. 299-337.
Berlucchi G. Learning and interheroispheric transfer of visual pattern discrimination in split chiasm cats / G. Berlucchi, J.M. Spraque, A. Antonini, A. Simoni // Exp. Brain Res. — 1979. — Vol. 34, № 5. — P. 551-574.
Berlucchi, J.M. Spraque, A. Antonini, A. Simoni // Exp. Brain Res. — 1979. — Vol. 34, № 5. — P. 551-574.
Myers J.J. Interhemispheric communication after section of the forebrain comissures / J.J. Myers, R.W. Sperry // Cortex. — 1985. — Vol. 21, № 2. — P. 249-260.
Geffen G. Interhemispheric control of manual motor activity / G. Geffen, D. Jones, L. Geffen // Behavioral Brain Research. — 1994. — Vol. 64, № 1. -P. 131-140.
Merola J.L. The effect of task difficulty upon the extent to which performance benefits from between-hemisphere division of inputs / J.L. Merola, J. Liederman // Intern. J. of Neuroscience. — 1990. — Vol. 54, № 1. — P. 35-44.
Banich M.T. Interhemispheric interaction: How do the two hemispheres divide and conquer a task / M.T. Banich, A. Belger // Cortex. — 1990. -Vol. 26, № 1. — P. 77-94.
Sereno A.B. Discrimination within and between hemifields: A new constraint on theories of attention / A. B. Sereno, S.M. Kosslyn // Neuropsychologia. — 1991. — Vol. 29, № 7. — P. 659-675.
B. Sereno, S.M. Kosslyn // Neuropsychologia. — 1991. — Vol. 29, № 7. — P. 659-675.
Thatcher R.W. Human cerebral hemispheres develop at different rates and ages / R.W. Thatcher, R.A Walker, S. Guidice // Science. — 1987. -Vol. 236.-P. 1110-1113.
Газзанига М. Расщепленный человеческий мозг / M. Газзанига // Хрестоматия по нейропсихологии / Под ред. Е.Д. Хомской. — М.: Изд-во РПО, 1999.-С. 128-132.
Bogen J.E. Split-brain syndromes / J.E. Bogen // Handbook of clinical neurology. Clinical neuropsychology / Ed. by J.A.M. Frederiks. — Amsterdam: Elsevier Publishing, 1985. — Vol. 1, № 45. — P. 99-106.
Московичюте Л.И. О роли мозолистого тела в организации высших психических функций / Л.И. Московичюте, Э.Г. Симерницкая, Н.А. Смирнов, Ю.Ф. Филатов // А.Р. Лурия и современная психология / Под ред. Е.Д. Хомской и др. — М.: Изд-во МГУ, 1982. — С. 143-150.
Симерницкая Э.Г. Нейропсихологическая диагностика нарушений памяти при поражениях мозолистого тела / Э. . — P. 275-282.
. — P. 275-282.
Corballis M. Interhemispheric visual integration in tfiree cases of familial callosal agenesis / M. Corballis, D. Finlay // Neuropsychology. — 2000. -Vol. 14, №1.-P. 60-70.
Egaas B. Redused size of CC autism / B. Egaas, E. Courchesne, O. Saitoh // Archives of Neurology. — 1995. — Vol. 52, № 8. — P. 794-801.
David A.S. Callosal transfer in schizophrenia: Too much or too little? / A.S. David // Journal of Abnormal Psychology. — 1993. — Vol. 102, № 4. — P. 573-579.
Орлова B.A. Морфологические особенности мозолистого тела в семьях больных шизофренией (по данным МРТ-исследования) / В.А. Орлова, В.И. Трубников, Н.Ю. Саватеева и др. // Социальная и клиническая психиатрия. — 2000. — Т. 10, № 3. — С. 6-9.
Hynd G. Corpus callosum morphology in attention deficit hyperactivity disorder / G. Hynd, M. Semrud-Clikemen, A. Lorys, E. Novey, D. Eliopulos // Journal of Learning Disabilities.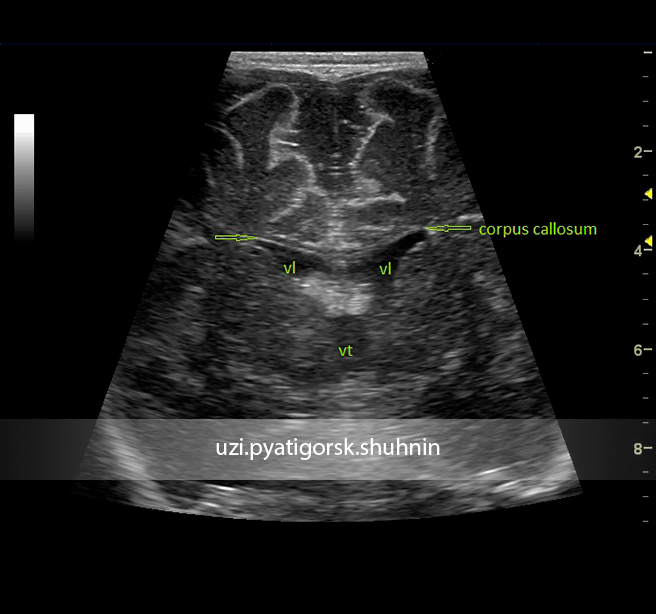 — 1991. — Vol. 24, № 3. — P. 141-145.
— 1991. — Vol. 24, № 3. — P. 141-145.
Njiokiktjien S. Callosal size in children with learning disabilities / S. Njiokiktjien, L. de Sonneville, G. Vaal // Behaviaral Brain Research. — 1994. — Vol. 64, № 1-2. — P. 213-218.
Hynd G. Dyslexia and CC morphology / G. Hynd, J. Hall, E. Novey, D. Eliopulos // Archives and neurology. — 1995. — Vol. 52, № 1. — P. 32-38.
Markee T. Callosal function in dyslexia: evoked potential inter-hemispheric transfer time and bilateral field advantage / T. Markee, W. Brown, L. Moore, D. Theberge // Developmental Neuropsychology. — 1996. — Vol. 12, № 4. — P. 409-428.
Chiarello С A house divided? Cognitive functioning with callosal agenesis / С Chiarello // Brain and Language. — 1980. — Vol. 11, № 1. — P. 128- 158.
Banich M. A lifespan perspective on interaction between the cerebral hemispheres / M. Banich, W. Brown // Developmental Neuropsychology. -2000.-Vol. 18, № 1.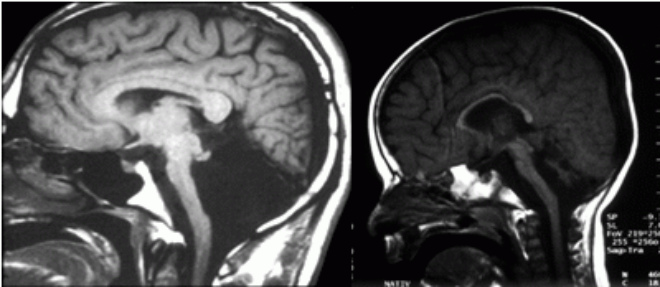 — P. 1-10.
— P. 1-10.
Banich M. Interhemispheric interaction during childhood: Neirologically intact children / M. Banich, A. Passarotti, D. Janes // Developmental Neuropsychology. — 2000. — Vol. 18,№ 1.-P. 33-51.
Медведев М.И. Клинические проявления и нейрорадиологические признаки агенезии мозолистого тела у новорожденного / М.И. Медведев, Н.Н. Володин, А.В. Горбунов и др. // Российский вестник перинатологии и педиатрии. — 2001. — Т. 46, № 5. — С. 56-58.
Ettlinger G. Agenesis of the corpus callosum: a further behavioral investigation / G. Ettlinger, С. Blakemore, A. Milner, J. Wilson // Brain. — 1974. — Vol. 97, № 1. — P. 225-234.
Jeret J.S. Clinicopathological findings associated with agenesis of the corpus callosum / J.S. Jeret, D. Serur, K..E. Wisniewski, R.A. Lubin // Brain Dev. — 1987. — Vol. 9, № 3. — P. 255-264.
Paul L. Communicative deficits in agenesis of the corpus callosum: Nonliteral language and affective prosody / L. Paul, D. Van Lancker-Sidtis, B. Schieffer et al. // Brain and Language. — 2003. — Vol. 85, № 2. — P. 313-324.
Kinsbourne M. Latency of uncrossed and of crossed reaction in callosal agenesis / M. Kinsbourne, M. Fisher // Neuropsychologia. — 1971. — Vol. 9, № 4. — P. 471-473.
Field M. Agenesis of the corpus callosum: report of two рге-school children and review of the literature / M. Field, R. Ashton, K. White // Developmental Medicine and Child Neurology. — 1978. — Vol. 20, № 1. — P. 47-61.
Koeda T. Tactile naming disorder of the left hand in two cases with corpus callosum agenesis / T. Koeda, K. Takeshita // Developmental Medicine and Child Neurology. — 1993. — Vol. 35, № 1. — P. 65-69.
Meerwaldl JD. Disturbunces of spatial perception in a patient with agenesis of the corpus callosum / JD. Meerwaldt // Neuropsychologia. — 1983. -Vol. 21, № 2. — P. 161-165.
Sauenvein H.C. Cognitive and sensori-motor functionning in the absance of corpus callosum: Neuropsychological studies in callosal agenesis and callosotomised patients / H. C. Sauenvein, M. Lassonde // Behav. Brain Res. — 1994. — Vol. 64, № 1-2. — P. 229-240.
C. Sauenvein, M. Lassonde // Behav. Brain Res. — 1994. — Vol. 64, № 1-2. — P. 229-240.
Fisher G. Mechanism of interhemispheric transfer and patterns of cognitive function in acallosal patients of normal intelligence / G. Fisher, S. Ryan, W. Dobyns // Archives of Neurology. — 1992. — Vol. 49, № 3. — P. 271-277.
Callosal agenesis: Natural split brain? / Ed. by M. Lassonde, M. Jeeves. — N.Y.: Plenum Press, 1994. — 307 p.
Friefeld S. A Comparative study of inter- and intrahemispheric somatosensory functions in children with partial and complete agenesis of the corpus callosum / S. Friefeld, D. MacGregor, S. Chuang, J. Saint-Cyr // Developmental Medicine and Child Neurology. — 2000. — Vol. 42, № 12. — P. 831-838.
Sullivan M.V. Memory Establishment via the anterior commissure of monkeys / M.V. Sullivan, C.R. Hamilton // Physiology and Behavior. — 1973. — Vol. 11, № 9. — P. 873-879.
Black P. Visual function of the forebrain commissures in the chimpanzee / P. Black, R.E. Myers // Science. — 1964. — Vol. 146, № 6. — P. 799-800.
Black, R.E. Myers // Science. — 1964. — Vol. 146, № 6. — P. 799-800.
Rauch R.A. Magnetic resonance imaging of corpus callosum Dysgenesis / R.A. Rauch, J.R. Jinkins // Callosal agenesis: Natural split brain? / Ed. by M. Lassonde, M. Jeeves. — N.Y.: Plenum Press, 1994. — P. 83-96.
Martin A. A qualitative limitation on visual transfer via the anterior commissure / A. Martin // Brain. — 1985. — Vol. 108, № 1. — P. 43-63.
Gazzaniga M. Profiles of right-hemisphere language and speech following brain bisection / M. Gazzaniga, С Smylie, K. Baynes, W. Hirst, С McClearly // Brain and Language. — 1984. — Vol. 22, № 2. — P. 206-220.
Brown W. Bilateral field advantage and evoked potential interhemispheric transmission in commissurotomy and callosum agenesis / W. Brown, M. Jeeves, R. Dietrich, D. Burnison // Neuropsychologia. — 1999. — Vol. 37, № 10. — P. 1165-1180.
Brown W.S. Interhemispheric Stroop effects in partial and complete agenesis of the corpus callosum / W. S. Brown, E.D. Thrasher, L.K. Paul // J. Int. Neuropsychol. Soc. — 2001. — Vol. 7, № 3. — P. 302-311.
S. Brown, E.D. Thrasher, L.K. Paul // J. Int. Neuropsychol. Soc. — 2001. — Vol. 7, № 3. — P. 302-311.
PoirierP. Sound localization in acallosal human listeners / P. Poirier, S. Miljours, M. Lassonde, F. Lepore//Brain. — 1993. -Vol. 116, № l.-P. 53- 69.
Aglioti S. Hemispheric control of unilateral and bilateral responses to lateralised light stimuli after callosotomy and in callosal agenesis / S. Aglioti, G. Berlucchi, R. Pallini et al. // Exp. Brain Res. — 1993. — Vol. 95, № 1. — P. 151-165.
Silver P.H. Motor coordination in callosal agenesis / P.H. Silver, M.A. Jeeves // Callosal agenesis: Natural split brain? / Ed. by M. Lassonde, M.A. Jeeves. -N.Y.: Plenum Press, 1994. -P. 207-219.
Gladstone M. Anomalous bimanual coordination among dyslexic boys / M. Gladstone, C.T. Best, R.J. Davidson // Developmental Psychology. -1989. — Vol. 25, № 2. — P. 236-246.
Moore L.H. Bimanual coordination in the dyslexic adults / L. H. Moore, W.S. Brown, Т.Е. Markee et al. // Neuropsyhologia. — 1995. — Vol. 33, №6.-P. 781-793.
H. Moore, W.S. Brown, Т.Е. Markee et al. // Neuropsyhologia. — 1995. — Vol. 33, №6.-P. 781-793.
Serrien D. Role of the corpus callosum in the bimanual coordination: a comparison of patients with congenital and acquired callosal damage / D.Serrien, A. Nirkko, M. Wiesendanger // European Journal of Neuroscience. — 2001. — Vol. 14, № 11. — P. 1897-1905.
Watson R.T. Callosal apraxia / R.T. Watson, K.M. Heilman // Brain. — 1983. — Vol. 106, № 2. — P. 391-403.
Lausberg H. Pantomime to visual presentation of objects: left hand despraxia in patients with complete callosotomy / H. Lausberg, R.F. Cms, S. Kita, E. Zaidel, A. Ptito // Brain. — 2003. — Vol. 126, № 2. — P. 343-360.
Moutard L. Agenesis of corpus callosum: prenatal diagnosis and prognosis / L. Moutard, V. Kieffer, J. Feingold, A. Adamsbaum et al. // Child’s Nervous System. — 2003. — Vol. 19, № 7-8. — P. 471-476.
Brown W. Cognitive and psychosocial deficit in agenesis of the corpus callosum with normal intelligence / W. Brown, L. Paul // Cognitive Neuropsychiatry. — 2000. — Vol. 5, № 2. — P. 135-157.
Brown, L. Paul // Cognitive Neuropsychiatry. — 2000. — Vol. 5, № 2. — P. 135-157.
Sanders R. Sentence comprehension following agenesis of the corpus callosum / R. Sanders // Brain and Language. — 1989. — Vol. 37, № 1. — P. 59- 72.
Temple CM. Reading in callosal agenesis / CM. Temple, M.A. Jeeves, O.O. Vilarroya // Brain and Language. — 1990. — Vol. 39, № 2. — P. 235-253.
O’Brien G. The behavioral and developmental consequences of corpus callosal agenesis and Aicardi Syndrome / G. O’Brien // Callosal agenesis: Natural split brain? / Ed. by M. Lassonde, M. Jeeves. — N.Y.: Plenum Press, 1994. — P. 235-246.
TenHouten W.D. Alexithymia and the split brain: IV Gottschalk-Gleser contentanalysis, in overview / W.D. TenHouten, K.D. Hoppe, G.E. Bogen, D.O. Walter // Psychotherapy and Psychosomatics. — 1986. — Vol. 44, № 3. — P. 113-121.
Buchanan D. A proposed neuropsychological Basis of alexithymia / D. *11.
*11.
Browneli H.H. Surprise but not coherence: sensitivity to verbal humor in right-hemisphere patients / H.H. Browneli, D. Michel, J. Powelson, H. Gardner // Brain and Lang. — 1983. — Vol. 18, № 1. — P. 20-27.
Shammi P. Humour appreciation: a role of the right frontal lobe / P. Shammi, D.T. Stuss // Brain. — 1999. — Vol. 122, № 4. — P. 657-666.
Shammi P. The effects of normal aging on humor appreciation / P. Shammi, D.T. Stuss // J. Int. Neuropsychol. Soc. — 2003. — Vol. 9, № 6. — P. 855- 863.
Brown W.S. Comprehension of humor in primary agenesis of the corpus callosum / W.S. Brown, L.K. Paul, M. Symington, R. Dietrich // Neuropsychologia. — 2005. — Vol. 43, № 6. — P. 906-916.
Paul L.K. Social processing deficits in agenesis of the corpus callosum: narratives from the Thematic Appreciation Test / L.K. Paul, B. Schieffer, W.S. Brown // Arch. Clin. Neuropsychol. — 2004. — Vol. 19, № 2. — P..jpg) 215-225.
215-225.
Величко М.А. Особенности психических процессов у детей с врожденной патологией мозолистого тела (обзор литературы) / М.А. Величко, М.С. Ковязина // Особый ребенок. Исследования и опыт помощи. — 1998. — № 1. — С. 27-34.
Ковязина М.С. Особенности межполушарного взаимодействия в двигательной сфере у детей в норме и при отклонениях в развитии / М.С. Ковязина, Е.Ю. Балашова, М.С. Казакова // Журнал прикладной психологии. — 2005. — № 2-3. — С. 2-11.
Буклина С.Б. Мозолистое тело, межполушарное взаимодействие и функции правого полушария мозга / С.Б. Буклина // Журнал неврологии и психиатрии. — 2004. — № 5. — С. 8-14.
Клинический случай аплазии мозолистого тела у ротвейлера, сходный с синдромом Айкарди у людей
Людмила Коникова ([email protected]), Ольга Дубовицкая, ветеринарная клиника «Белый Клык» — отделение неврологии, Москва
Лекторы NVC
Описано два клинических случая патологии мозолистого тела у собак различной степени и этиологии..jpg) Приведены литературные данные по заболеванию, описаны характерные симптомы, методы диагностики и возможные сочетанные патологии других органов и систем. Высказано предположение о возможных генетических пороках развития. Проведена аналогия с синдромом Айкарди у людей.
Приведены литературные данные по заболеванию, описаны характерные симптомы, методы диагностики и возможные сочетанные патологии других органов и систем. Высказано предположение о возможных генетических пороках развития. Проведена аналогия с синдромом Айкарди у людей.
Введение
Мозолистое тело (corpus calossum) представляет собой сплетение 250 пучков нервных волокон и является самой большой структурой головного мозга плацентарных животных, соединяющей правое и левое полушария. К функциям мозолистого тела относится: связь между полушариями, участие в процессах осморегуляции, координация информации и обмен сенсорными стимулами между полушариями в процессе обучения и памяти.
Мозолистое тело может быть визуализировано на МРТ с 8-недельного возраста. Оно имеет гиперинтенсивный сигнал на Т1 последовательности и гипоинтенсивный сигнал на Т2 взвешенных МР-изображениях.
Наиболее информативными для исследования мозолистого тела являются сагиттальные срезы [6].
Волокна в мозолистом теле проходят главным образом в поперечном направлении, связывая симметричные участки противоположных полушарий. Однако некоторые волокна связывают и несимметричные места противоположных полушарий, например, лобные извилины с теменными или затылочными, а также разные участки одного полушария (так называемые ассоциативные пути).
К аномалиям мозолистого тела относят гипоплазию, гипоплазию с дисплазией и агенезию (аплазию).
Наиболее общим симптомом у собак при патологиях мозолистого тела является адипсия или гиподипсия [3]. Адипсическая гипернатриемия характеризуется хроническими и острыми эпизодами повышения натрия в крови, связанными с потерей жажды [3]. В патогенезе жажды у собак наибольшую роль играют рецепторы к ионам натрия, локализованные в краниальной части третьего желудочка, включая околожелудочковый орган, субфорникальный орган, срединные ядра, сосудистый орган концевой пластины [4].
Описание клинических случаев
1. Клинический случай аплазии мозолистого тела
В клинику «Белый Клык» поступил 6-месячный щенок породы ротвейлер по поводу тонических генерализованных приступов с потерей сознания, адипсии, эпизодической вялости, гипорексии, нарушения координации. При осмотре также зафиксирована прогнатия, микрофтальмия, недоразвитие нижней челюсти. Неврологического дефицита не выявлено. Общеклинический и биохимический анализ без особенностей. В электролитном профиле выявлена гипернатриемия (Na+ 173 ммоль/л (138–164), Cl– 129 ммоль/л (93–119)).
Гипернатриемия (более 170 ммоль/л у собак) [7] может быть причиной судорог с потерей сознания из-за дегидратации клеток нервной системы и отёка ткани головного мозга, что и было зафиксировано в данном случае. Собаке было проведено магнитно-резонансное исследование головного мозга.
Заключение МРТ:
Подозрение на аплазию мозолистого тела..jpg)
Лечение
Благодаря симптоматической терапии и элементарной диете с добавлением воды удалось добиться стабилизации состояния и профилактировать судороги у собаки. В данный момент состояние собаки удовлетворительное, приступов больше не отмечено.
2. Клинический случай гипоплазии с дисплазией мозолистого тела в сочетании с гидроцефалией
Собака породы йоркширский терьер, 3-месячного возраста, была доставлена в клинику «Белый Клык» с жалобами на выраженное нарушение координации и сознания (сопор), а также отсутствие жажды (адипсию). На неврологическом осмотре выявлен дефицит черепных нервов.
Топический диагноз: центральный вестибулярный синдром (ЦВС). УЗИ головного мозга выявило значительную вентрикуломегалию (более 60%) и повышенный индекс резистентности.
Собака была эвтаназирована по решению владельцев с предварительным диагнозом: «Первичная гидроцефалия. Гипоплазия мозолистого тела».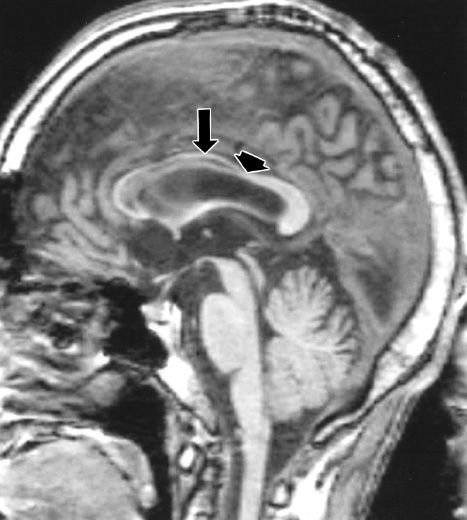
На вскрытии было зафиксировано выраженное недоразвитие костных структур черепа и множественные патологии головного мозга: атрофия всех структур головного мозга, включая сагиттальный венозный синус, выраженная вентрикуломегалия, гипоплазия с дисплазией мозолистого тела, гипоплазия мозжечка.
Обсуждение
Сообщалось о связи между ранней манифестацией болезни лизосомальных накоплений (ганглиозидоз) и гипоплазии мозолистого тела. Связь между патологией мозолистого тела и врождёнными нарушениями метаболизма установлена у людей.
Это позволяет думать, что на молекулярном уровне развитие мозолистого тела вовлекает синхронизацию многих клеточных процессов.
Нарушение этих механизмов лежит в основе полной или частичной потери мозолистого тела. К сожалению, у собак нет генетических тестов на болезни лизосомальных накоплений.
Сочетание аплазии мозолистого тела, лицевого дисморфизма, аномалии развития глаз, а также нарушение развития костных структур (клинический случай №1) имеет сходство с синдромом Айкарди у людей — генетическим заболеванием, характеризующимся частичным или полным отсутствием одной из основных структур мозга — мозолистого тела и пороками развития других органов [5].
Заключение
Существуют различные степени мальформации мозолистого тела, в случаях гипоплазии у собак мозолистое тело наиболее повреждено рострально [3]. Собаки с мальформацией в области мозолистого тела демонстрируют адипсию или гиподипсию, а также связанную с этим гипернатриемию. Патологии мозолистого тела обычно сочетаны с другими мальформациями и аномалиями строения. В первом клиническом случае отмечено довольно редкое для собак сочетание лицевого дисморфизма и аплазии мозолистого тела.
В связи с тем, что врождённые аплазии мозолистого тела —
довольно редкое явление у собак, необходимы дальнейшие научные и сравнительные исследования на данную тему с большей выборкой пациентов для получения более точных статистических данных.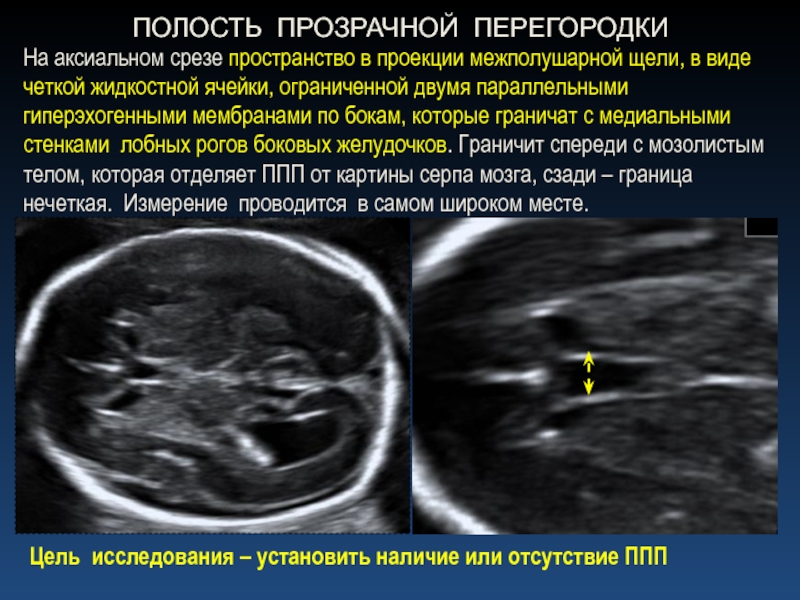
Литература
1. Raybaud, C. & Di Rocco. The corpus callosum, the other great forebrain commissures, and the septum pellucidum: anatomy, development, and malformation. Neuroradiology, Vol. 52, No. 6, (June 2010), pp. 447–77, ISSN 0028-3940 РC. (2007).
2. Gross B.Garcia-Tapia D, Riedisel E. Et normal canine brain maturation at MRI. Vet Radiol Ultrasound 2010;51:361–373.
3. R.Goncalves, H Volk, P.M Smith, J.Penderis — Corpus colossal Abnormalities in Dogs — J vet Intern Med 2014;28:1275–1279
4. Antunes-Rodrigues J. de Castro M. Elias LL.et al. Neuroendocrine control of body fluid metabolism. Physiol Rev 2004;84:169–208
5. Uggetti, C et al. «Aicardi-Goutieres syndrome: neuroradiologic findings and follow-ups». AJNR Am J Neuroradiol 2009; 30.
6. Harding BN, Copp AJ. Malformations. In: Graham DI, Lantons PL (eds): Greenfield’s neuropathology. London: Arnold, 2002; 357–470.
7. Luisa De Risio, Simon Platt. Canine and Feline Epilepsy Diagnosis and Management. 2014; 64–65.
Canine and Feline Epilepsy Diagnosis and Management. 2014; 64–65.
СВМ № 3/2015
Оценить материал
НравитсяНравится Поздравляю Сочувствую Возмутительно Смешно Задумался Нет слов
Агенезия мозолистого тела — NORD (Национальная организация по редким заболеваниям)
Агенезия мозолистого тела (ОСС) может первоначально проявиться в начале эпилептических припадков в течение первых недель жизни или в течение первых двух лет. Однако не у всех людей с АКК случаются судороги. (Для получения дополнительной информации об этих типах припадков выберите «эпилепсия» в качестве поискового запроса в базе данных редких заболеваний).
Другими симптомами, которые могут проявляться в раннем возрасте, являются проблемы с кормлением и задержка в удерживании головы прямо. Сидение, стояние и ходьба также могут задерживаться. Нарушение умственного и физического развития и / или скопление жидкости в черепе (гидроцефалия) также являются симптомами раннего начала этого заболевания. (Для получения дополнительной информации выберите «гидроцефалия» в качестве поискового запроса в базе данных редких заболеваний.)
Сидение, стояние и ходьба также могут задерживаться. Нарушение умственного и физического развития и / или скопление жидкости в черепе (гидроцефалия) также являются симптомами раннего начала этого заболевания. (Для получения дополнительной информации выберите «гидроцефалия» в качестве поискового запроса в базе данных редких заболеваний.)
Непрогрессирующая умственная отсталость, нарушение зрительно-моторной координации и нарушение зрительной или слуховой (слуховой) памяти можно диагностировать с помощью неврологического тестирования пациентов. с ACC.
В некоторых легких случаях симптомы могут не проявляться в течение многих лет. Пациентам пожилого возраста обычно ставят диагноз во время тестов на такие симптомы, как судороги, монотонная или повторяющаяся речь или головные боли. В легких случаях на это можно не обращать внимания из-за отсутствия явных симптомов в детстве.
У некоторых пациентов могут быть глубоко посаженные глаза и выступающий лоб. Может присутствовать аномально маленькая голова (микроцефалия), а иногда и необычно большая голова (макроцефалия). Кожные пятна на коже перед ушами (предурикулярные кожные метки), один или несколько согнутых пальцев (камптодактилия) и задержка роста также были связаны с некоторыми случаями агенезии мозолистого тела.В других случаях широко расставленные глаза (телекантус), небольшой нос с поднятыми (антевертированными) ноздрями, уши неправильной формы, чрезмерная кожа шеи, короткие руки, снижение мышечного тонуса (гипотония), аномалии гортани, пороки сердца и симптомы Может присутствовать синдром Пьера-Робена. (Для получения дополнительной информации выберите «Pierre-Robin» в качестве поискового запроса в базе данных редких заболеваний).
Кожные пятна на коже перед ушами (предурикулярные кожные метки), один или несколько согнутых пальцев (камптодактилия) и задержка роста также были связаны с некоторыми случаями агенезии мозолистого тела.В других случаях широко расставленные глаза (телекантус), небольшой нос с поднятыми (антевертированными) ноздрями, уши неправильной формы, чрезмерная кожа шеи, короткие руки, снижение мышечного тонуса (гипотония), аномалии гортани, пороки сердца и симптомы Может присутствовать синдром Пьера-Робена. (Для получения дополнительной информации выберите «Pierre-Robin» в качестве поискового запроса в базе данных редких заболеваний).
Синдром Айкарди, который, как считается, наследуется как Х-сцепленное доминантное заболевание, состоит из агенеза мозолистого тела, детских спазмов и аномального строения глаза.Это чрезвычайно редкое врожденное заболевание, при котором частые судороги, разительные аномалии средней оболочки глаза (сосудистой оболочки) и слоев сетчатки, а также отсутствие структуры, соединяющей два полушария головного мозга (мозолистое тело), сопровождают тяжелую умственную отсталость.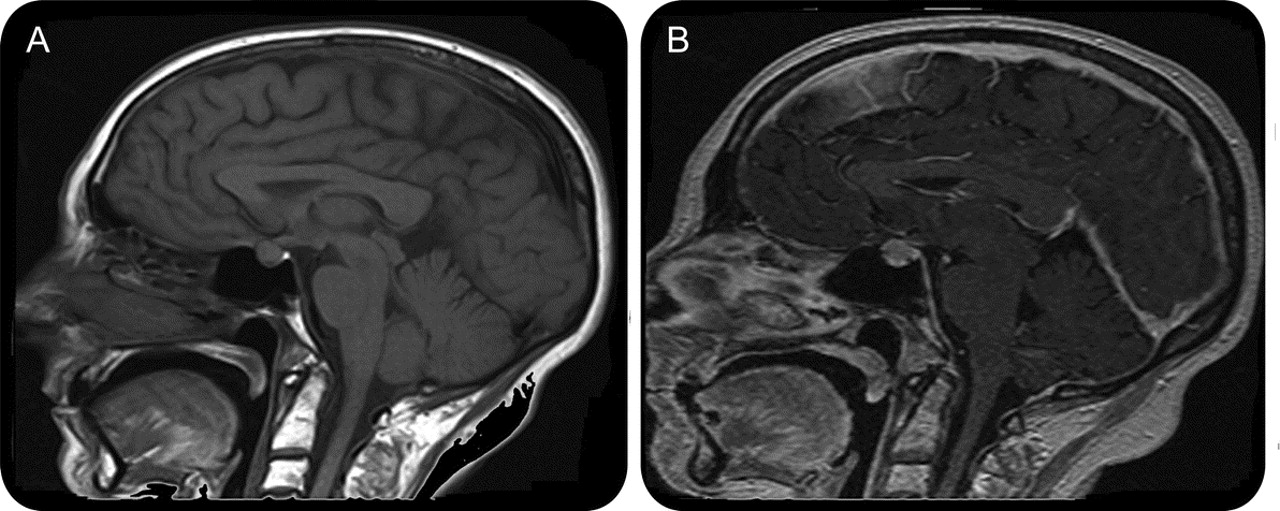 Поражаются только самки. (Для получения дополнительной информации об этом заболевании выберите «Айкарди» в качестве поискового запроса в базе данных редких заболеваний).
Поражаются только самки. (Для получения дополнительной информации об этом заболевании выберите «Айкарди» в качестве поискового запроса в базе данных редких заболеваний).
Синдром Андермана, выявленный в 1972 году, представляет собой генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела, умственной отсталости и прогрессирующих нарушений сенсомоторной нервной системы (нейропатия).Все известные случаи этого заболевания происходят из округа Шарлевуа и района Сагеней-Лак-Сен-Жан в Квебеке, Канада. Ген, вызывающий эту редкую форму АСС, был недавно идентифицирован, и в настоящее время доступно тестирование этого гена (SLC12A6).
XLAG (X-связанная лиссэнцефалия с неоднозначными гениталиями — редкое генетическое заболевание, при котором у мужчин маленький и гладкий мозг (лиссэнцефалия), маленький пенис, тяжелая умственная отсталость и трудноизлечимая эпилепсия. Это вызвано мутациями в гене ARX.У женщин эти же мутации могут вызывать только ACC, тогда как менее серьезные мутации у мужчин могут вызывать умственную отсталость. Тестирование на это заболевание также доступно клинически.
Тестирование на это заболевание также доступно клинически.
Детская агенезия мозолистого тела — условия и методы лечения
В этой секции Подробнее по этой теме
Обновление по коронавирусу: Что нужно знать пациентам и их семьям Закрыть оповещениеЧто такое агенезия мозолистого тела?
Мозолистое тело является частью головного мозга.Его нервные волокна соединяют две стороны (полушария головного мозга) мозга. Агенезия мозолистого тела — это врожденный дефект, который возникает, когда эта структура не развивается должным образом.
Для детей, рожденных с агенезией мозолистого тела, существует широкий диапазон исходов, от нормального функционирования в самых легких случаях до ряда потенциальных проблем со здоровьем и развитием по мере увеличения степени тяжести.
Узнайте больше о нашем Институте пренатальной педиатрии.
 js»>
js»>Во многих случаях мы не можем определить причину агенезии мозолистого тела. Известные причины включают:
- Хромосомные дефекты, влияющие на развитие мозга плода
- Определенные вирусные инфекции у матери, которые передаются развивающемуся ребенку
- Воздействие на будущего ребенка определенных токсинов или лекарств
- Аномальное развитие мозга, вызванное кистами
Если нет других аномалий развития мозга, у младенцев может быть немного, если таковые имеются, в более поздней жизни.Новорожденные с агенезом мозолистого тела могут не проявлять симптомы сразу, особенно если у них нет других сопутствующих заболеваний. Общие симптомы, которые могут стать более очевидными в младенчестве и детстве, включают:
- Судороги
- Когнитивные нарушения
- Плохое питание и трудности с глотанием
- Задержка развития двигательных и языковых навыков, таких как сидение, ходьба и разговор
- Зрение и слух нарушение
- Плохой мышечный тонус и координация
- Бессонница или другие проблемы со сном
- Психосоциальные трудности, включая синдром дефицита внимания (СДВ), навязчивое поведение и социальную незрелость
- Когнитивные трудности, включая нарушение обучаемости и проблемы с абстрактным мышлением и решением проблем
Ваш врач может заподозрить агенезию мозолистого тела на основании обычных пренатальных ультразвуковых исследований, проводимых во время беременности. Ваш врач может направить вас в наш институт пренатальной педиатрии для дальнейшего обследования, чтобы оценить состояние вашего ребенка. Тесты могут включать:
Ваш врач может направить вас в наш институт пренатальной педиатрии для дальнейшего обследования, чтобы оценить состояние вашего ребенка. Тесты могут включать:
- Ультразвук высокого разрешения (уровень II) для подтверждения диагноза
- МРТ плода, чтобы помочь подтвердить диагноз и определить, присутствуют ли связанные с ним проблемы с мозгом. Этот расширенный визуализирующий тест с высоким разрешением дает наилучшую информацию об основной причине и лучшее изображение остальной части мозга ребенка.
- МРТ после рождения для получения дополнительной информации о вашем ребенке
- Поскольку агенезия мозолистого тела может быть связана с хромосомными или генетическими аномалиями, мы можем порекомендовать генетическое тестирование с помощью амниоцентеза во время беременности или анализов крови ребенка после рождения.
Узнайте больше об услугах дородового ухода, связанных с повышенным риском, в Национальном институте пренатальной педиатрии.
В настоящее время не существует методов восстановления мозолистого тела до нормального состояния.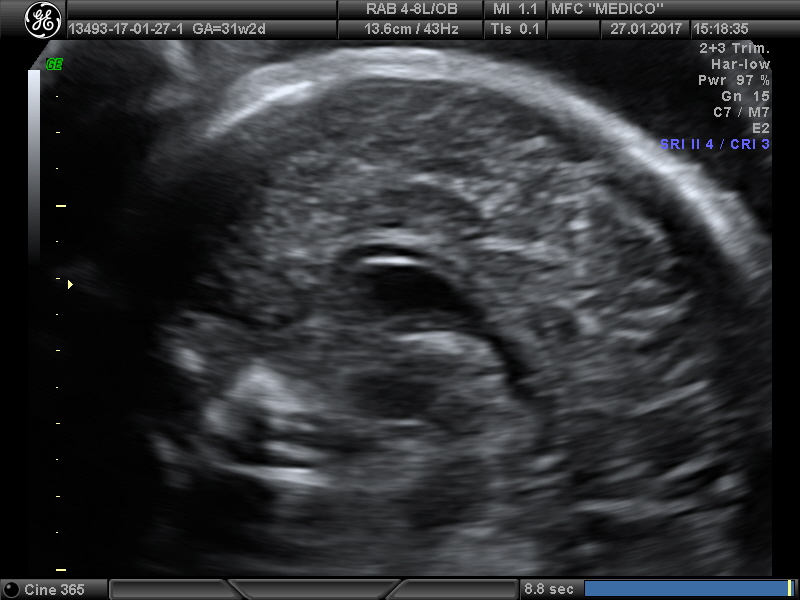 Основной курс лечения агенезии мозолистого тела — это устранение возможных осложнений. Варианты лечения могут включать:
Основной курс лечения агенезии мозолистого тела — это устранение возможных осложнений. Варианты лечения могут включать:
- Лекарства для контроля припадков
- Речевая терапия для помощи в развитии речи и языка
- Физическая терапия для улучшения мышечной силы и координации
- Трудотерапия для саморазвития навыки ухода и мобильности, такие как прием пищи, одевание и ходьба
- Специальное образование , необходимое для когнитивных проблем и проблем с обучением
Неврологи и нейрохирурги, участвующие в программе дородового исследования мозга в нашем институте пренатальной педиатрии, имеют большой опыт в лечении агенеза мозолистого тела.Мы разрабатываем планы, обеспечивающие непрерывность ухода на ранних стадиях беременности, после родов и на протяжении всего детства.
История Сэди
Сэди — одна из сотен младенцев с острой энцефалопатией, перенесших терапевтическую гипотермию всего тела в Национальном детском отделении интенсивной терапии новорожденных. Лечебная гипотермия всего тела предлагается в первые несколько часов жизни, чтобы сохранить клетки мозга и предотвратить инвалидность или смерть.
Лечебная гипотермия всего тела предлагается в первые несколько часов жизни, чтобы сохранить клетки мозга и предотвратить инвалидность или смерть.
Дайте действовать
Поделитесь своим днем рождения с ребенком. Отпразднуйте свою жизнь и дайте шанс тому, кто отчаянно хочет иметь столько же, сколько вы.
Сделай этоМозолистое тело, Агенезия: причины, симптомы и лечение
Мозолистое тело — это структура, которая соединяет правую и левую части мозга. Он содержит 200 миллионов нервных волокон, которые передают информацию туда и обратно.
Агенезия мозолистого тела (АСС) — это врожденный дефект, который возникает, когда связи между правой и левой сторонами мозга ребенка не формируются правильно.По оценкам, это происходит у 1-7 из 4000 живорождений.
Существует несколько специфических форм ОСС, в том числе:
- частичная агенезия мозолистого тела
- гипоплазия мозолистого тела
- гипоплазия мозолистого тела
- дисгенезия мозолистого тела
Ребенок, рожденный с ОЦК, может выжить с условием. Однако это может вызвать задержку развития, которая может быть легкой или более серьезной.
Однако это может вызвать задержку развития, которая может быть легкой или более серьезной.
Например, ACC может вызвать задержку в развитии у ребенка моторных навыков, таких как сидение, ходьба или езда на велосипеде.Это потенциально может вызвать трудности с глотанием и кормлением. Плохая координация также часто встречается у детей с этим заболеванием.
У ребенка также могут наблюдаться задержки речи и речи при выразительном общении.
Хотя могут наблюдаться когнитивные нарушения, многие люди с ОКК обладают нормальным интеллектом.
Другие потенциальные симптомы ACC включают:
ACC — врожденный врожденный дефект. Это означает, что он присутствует при рождении.
Мозолистое тело ребенка формируется в конце первого триместра беременности.Множество факторов риска могут увеличить вероятность развития ОКК.
В течение первого триместра беременности воздействие определенных лекарств, таких как вальпроат, повышает риск развития АКК у ребенка. Воздействие определенных наркотиков и алкоголя во время беременности — еще один фактор риска.
Воздействие определенных наркотиков и алкоголя во время беременности — еще один фактор риска.
Если биологическая мать вашего ребенка заболевает определенными вирусными инфекциями во время беременности, например краснухой, это также может вызвать ОКК.
Хромосомные повреждения и аномалии также могут повышать риск ОКК у ребенка.Например, трисомия связана с ACC. При трисомии у вашего ребенка есть дополнительная копия хромосомы 8, 13 или 18.
Большинство случаев АСС возникает наряду с другими аномалиями головного мозга. Например, если кисты развиваются в головном мозге ребенка, они могут блокировать рост его мозолистого тела и вызывать ОКК.
Другие состояния также могут быть связаны с ACC, в том числе:
Некоторые из этих состояний вызваны генетическими нарушениями.
Если у вашего ребенка АКК, врач может обнаружить его еще до его рождения во время пренатального ультразвукового исследования.Если они видят признаки ОКС, они могут заказать МРТ для подтверждения диагноза.
В других случаях ППК вашего ребенка может оставаться незамеченным до его рождения. Если их врач подозревает, что у них ОКК, они могут назначить МРТ или КТ, чтобы проверить состояние.
Лекарства от ОКС не существует, но врач вашего ребенка может назначить лечение, чтобы помочь справиться с его симптомами.
Например, они могут порекомендовать лекарства для контроля припадков. Они также могут порекомендовать речевую, физическую или профессиональную терапию, чтобы помочь вашему ребенку справиться с другими симптомами.
В зависимости от тяжести состояния ваш ребенок может вести долгую и здоровую жизнь с ACC. Обратитесь к врачу за дополнительной информацией об их конкретном состоянии, вариантах лечения и долгосрочном прогнозе.
ACC — это врожденный дефект, который может вызывать задержку развития от легкой до тяжелой. Экологические и генетические факторы могут играть роль в его развитии.
Если у вас есть ребенок с АКК, его врач может порекомендовать лекарства, реабилитационную терапию или другие методы лечения, которые помогут справиться с его симптомами. Их врач может предоставить дополнительную информацию о вариантах лечения и долгосрочных перспективах.
Их врач может предоставить дополнительную информацию о вариантах лечения и долгосрочных перспективах.
Мозолистое тело, агенезия: причины, симптомы и лечение
Мозолистое тело — это структура, соединяющая правое и левое полушарие мозга. Он содержит 200 миллионов нервных волокон, которые передают информацию туда и обратно.
Агенезия мозолистого тела (АСС) — это врожденный дефект, который возникает, когда связи между правой и левой сторонами мозга ребенка не формируются правильно.По оценкам, это происходит у 1-7 из 4000 живорождений.
Существует несколько специфических форм ОСС, в том числе:
- частичная агенезия мозолистого тела
- гипоплазия мозолистого тела
- гипоплазия мозолистого тела
- дисгенезия мозолистого тела
Ребенок, рожденный с ОЦК, может выжить с условием. Однако это может вызвать задержку развития, которая может быть легкой или более серьезной.
Например, ACC может вызвать задержку в развитии у ребенка моторных навыков, таких как сидение, ходьба или езда на велосипеде..jpg) Это потенциально может вызвать трудности с глотанием и кормлением. Плохая координация также часто встречается у детей с этим заболеванием.
Это потенциально может вызвать трудности с глотанием и кормлением. Плохая координация также часто встречается у детей с этим заболеванием.
У ребенка также могут наблюдаться задержки речи и речи при выразительном общении.
Хотя могут наблюдаться когнитивные нарушения, многие люди с ОКК обладают нормальным интеллектом.
Другие потенциальные симптомы ACC включают:
ACC — врожденный врожденный дефект. Это означает, что он присутствует при рождении.
Мозолистое тело ребенка формируется в конце первого триместра беременности.Множество факторов риска могут увеличить вероятность развития ОКК.
В течение первого триместра беременности воздействие определенных лекарств, таких как вальпроат, повышает риск развития АКК у ребенка. Воздействие определенных наркотиков и алкоголя во время беременности — еще один фактор риска.
Если биологическая мать вашего ребенка заболевает определенными вирусными инфекциями во время беременности, например краснухой, это также может вызвать ОКК.
Хромосомные повреждения и аномалии также могут повышать риск ОКК у ребенка.Например, трисомия связана с ACC. При трисомии у вашего ребенка есть дополнительная копия хромосомы 8, 13 или 18.
Большинство случаев АСС возникает наряду с другими аномалиями головного мозга. Например, если кисты развиваются в головном мозге ребенка, они могут блокировать рост его мозолистого тела и вызывать ОКК.
Другие состояния также могут быть связаны с ACC, в том числе:
Некоторые из этих состояний вызваны генетическими нарушениями.
Если у вашего ребенка АКК, врач может обнаружить его еще до его рождения во время пренатального ультразвукового исследования.Если они видят признаки ОКС, они могут заказать МРТ для подтверждения диагноза.
В других случаях ППК вашего ребенка может оставаться незамеченным до его рождения. Если их врач подозревает, что у них ОКК, они могут назначить МРТ или КТ, чтобы проверить состояние.
Лекарства от ОКС не существует, но врач вашего ребенка может назначить лечение, чтобы помочь справиться с его симптомами.
Например, они могут порекомендовать лекарства для контроля припадков. Они также могут порекомендовать речевую, физическую или профессиональную терапию, чтобы помочь вашему ребенку справиться с другими симптомами.
В зависимости от тяжести состояния ваш ребенок может вести долгую и здоровую жизнь с ACC. Обратитесь к врачу за дополнительной информацией об их конкретном состоянии, вариантах лечения и долгосрочном прогнозе.
ACC — это врожденный дефект, который может вызывать задержку развития от легкой до тяжелой. Экологические и генетические факторы могут играть роль в его развитии.
Если у вас есть ребенок с АКК, его врач может порекомендовать лекарства, реабилитационную терапию или другие методы лечения, которые помогут справиться с его симптомами.Их врач может предоставить дополнительную информацию о вариантах лечения и долгосрочных перспективах.
Мозолистое тело, агенезия: причины, симптомы и лечение
Мозолистое тело — это структура, соединяющая правое и левое полушарие мозга. Он содержит 200 миллионов нервных волокон, которые передают информацию туда и обратно.
Он содержит 200 миллионов нервных волокон, которые передают информацию туда и обратно.
Агенезия мозолистого тела (АСС) — это врожденный дефект, который возникает, когда связи между правой и левой сторонами мозга ребенка не формируются правильно.По оценкам, это происходит у 1-7 из 4000 живорождений.
Существует несколько специфических форм ОСС, в том числе:
- частичная агенезия мозолистого тела
- гипоплазия мозолистого тела
- гипоплазия мозолистого тела
- дисгенезия мозолистого тела
Ребенок, рожденный с ОЦК, может выжить с условием. Однако это может вызвать задержку развития, которая может быть легкой или более серьезной.
Например, ACC может вызвать задержку в развитии у ребенка моторных навыков, таких как сидение, ходьба или езда на велосипеде.Это потенциально может вызвать трудности с глотанием и кормлением. Плохая координация также часто встречается у детей с этим заболеванием.
У ребенка также могут наблюдаться задержки речи и речи при выразительном общении.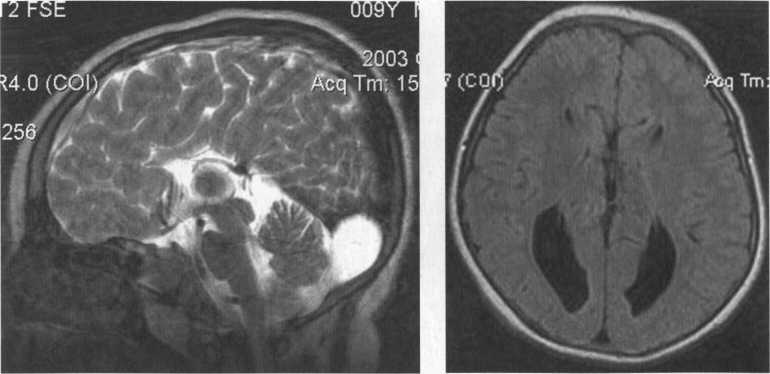
Хотя могут наблюдаться когнитивные нарушения, многие люди с ОКК обладают нормальным интеллектом.
Другие потенциальные симптомы ACC включают:
ACC — врожденный врожденный дефект. Это означает, что он присутствует при рождении.
Мозолистое тело ребенка формируется в конце первого триместра беременности.Множество факторов риска могут увеличить вероятность развития ОКК.
В течение первого триместра беременности воздействие определенных лекарств, таких как вальпроат, повышает риск развития АКК у ребенка. Воздействие определенных наркотиков и алкоголя во время беременности — еще один фактор риска.
Если биологическая мать вашего ребенка заболевает определенными вирусными инфекциями во время беременности, например краснухой, это также может вызвать ОКК.
Хромосомные повреждения и аномалии также могут повышать риск ОКК у ребенка.Например, трисомия связана с ACC. При трисомии у вашего ребенка есть дополнительная копия хромосомы 8, 13 или 18.
Большинство случаев АСС возникает наряду с другими аномалиями головного мозга. Например, если кисты развиваются в головном мозге ребенка, они могут блокировать рост его мозолистого тела и вызывать ОКК.
Другие состояния также могут быть связаны с ACC, в том числе:
Некоторые из этих состояний вызваны генетическими нарушениями.
Если у вашего ребенка АКК, врач может обнаружить его еще до его рождения во время пренатального ультразвукового исследования.Если они видят признаки ОКС, они могут заказать МРТ для подтверждения диагноза.
В других случаях ППК вашего ребенка может оставаться незамеченным до его рождения. Если их врач подозревает, что у них ОКК, они могут назначить МРТ или КТ, чтобы проверить состояние.
Лекарства от ОКС не существует, но врач вашего ребенка может назначить лечение, чтобы помочь справиться с его симптомами.
Например, они могут порекомендовать лекарства для контроля припадков. Они также могут порекомендовать речевую, физическую или профессиональную терапию, чтобы помочь вашему ребенку справиться с другими симптомами.
В зависимости от тяжести состояния ваш ребенок может вести долгую и здоровую жизнь с ACC. Обратитесь к врачу за дополнительной информацией об их конкретном состоянии, вариантах лечения и долгосрочном прогнозе.
ACC — это врожденный дефект, который может вызывать задержку развития от легкой до тяжелой. Экологические и генетические факторы могут играть роль в его развитии.
Если у вас есть ребенок с АКК, его врач может порекомендовать лекарства, реабилитационную терапию или другие методы лечения, которые помогут справиться с его симптомами.Их врач может предоставить дополнительную информацию о вариантах лечения и долгосрочных перспективах.
Мозолистое тело, агенезия: причины, симптомы и лечение
Мозолистое тело — это структура, соединяющая правое и левое полушарие мозга. Он содержит 200 миллионов нервных волокон, которые передают информацию туда и обратно.
Агенезия мозолистого тела (АСС) — это врожденный дефект, который возникает, когда связи между правой и левой сторонами мозга ребенка не формируются правильно. По оценкам, это происходит у 1-7 из 4000 живорождений.
По оценкам, это происходит у 1-7 из 4000 живорождений.
Существует несколько специфических форм ОСС, в том числе:
- частичная агенезия мозолистого тела
- гипоплазия мозолистого тела
- гипоплазия мозолистого тела
- дисгенезия мозолистого тела
Ребенок, рожденный с ОЦК, может выжить с условием. Однако это может вызвать задержку развития, которая может быть легкой или более серьезной.
Например, ACC может вызвать задержку в развитии у ребенка моторных навыков, таких как сидение, ходьба или езда на велосипеде.Это потенциально может вызвать трудности с глотанием и кормлением. Плохая координация также часто встречается у детей с этим заболеванием.
У ребенка также могут наблюдаться задержки речи и речи при выразительном общении.
Хотя могут наблюдаться когнитивные нарушения, многие люди с ОКК обладают нормальным интеллектом.
Другие потенциальные симптомы ACC включают:
ACC — врожденный врожденный дефект. Это означает, что он присутствует при рождении.
Это означает, что он присутствует при рождении.
Мозолистое тело ребенка формируется в конце первого триместра беременности.Множество факторов риска могут увеличить вероятность развития ОКК.
В течение первого триместра беременности воздействие определенных лекарств, таких как вальпроат, повышает риск развития АКК у ребенка. Воздействие определенных наркотиков и алкоголя во время беременности — еще один фактор риска.
Если биологическая мать вашего ребенка заболевает определенными вирусными инфекциями во время беременности, например краснухой, это также может вызвать ОКК.
Хромосомные повреждения и аномалии также могут повышать риск ОКК у ребенка.Например, трисомия связана с ACC. При трисомии у вашего ребенка есть дополнительная копия хромосомы 8, 13 или 18.
Большинство случаев АСС возникает наряду с другими аномалиями головного мозга. Например, если кисты развиваются в головном мозге ребенка, они могут блокировать рост его мозолистого тела и вызывать ОКК.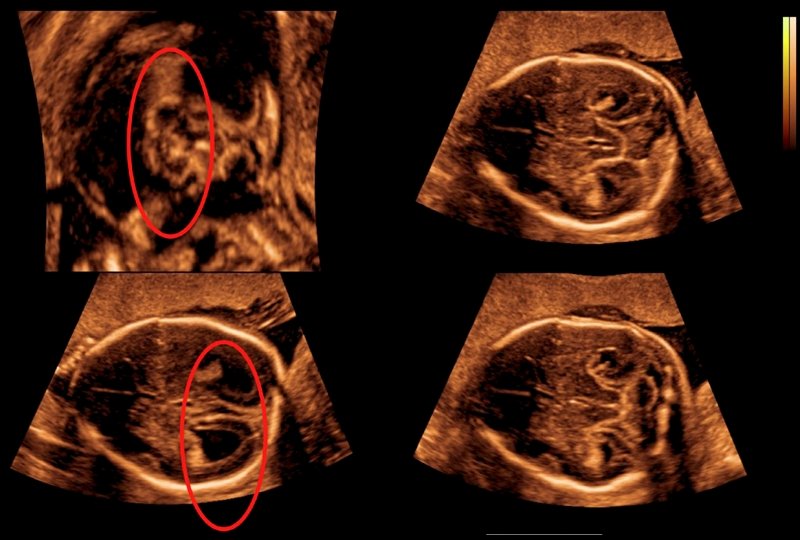
Другие состояния также могут быть связаны с ACC, в том числе:
Некоторые из этих состояний вызваны генетическими нарушениями.
Если у вашего ребенка АКК, врач может обнаружить его еще до его рождения во время пренатального ультразвукового исследования.Если они видят признаки ОКС, они могут заказать МРТ для подтверждения диагноза.
В других случаях ППК вашего ребенка может оставаться незамеченным до его рождения. Если их врач подозревает, что у них ОКК, они могут назначить МРТ или КТ, чтобы проверить состояние.
Лекарства от ОКС не существует, но врач вашего ребенка может назначить лечение, чтобы помочь справиться с его симптомами.
Например, они могут порекомендовать лекарства для контроля припадков. Они также могут порекомендовать речевую, физическую или профессиональную терапию, чтобы помочь вашему ребенку справиться с другими симптомами.
В зависимости от тяжести состояния ваш ребенок может вести долгую и здоровую жизнь с ACC. Обратитесь к врачу за дополнительной информацией об их конкретном состоянии, вариантах лечения и долгосрочном прогнозе.
ACC — это врожденный дефект, который может вызывать задержку развития от легкой до тяжелой. Экологические и генетические факторы могут играть роль в его развитии.
Если у вас есть ребенок с АКК, его врач может порекомендовать лекарства, реабилитационную терапию или другие методы лечения, которые помогут справиться с его симптомами.Их врач может предоставить дополнительную информацию о вариантах лечения и долгосрочных перспективах.
Исходы, связанные с изолированным агенезом мозолистого тела: метаанализ
Abstract
КОНТЕКСТ: Дородовое консультирование в случаях агенеза мозолистого тела (АКК) является сложной задачей.
ЗАДАЧИ: Определить исход у плодов с изолированной полной и частичной ОПП.
ИСТОЧНИКИ ДАННЫХ: базы данных Medline, Embase, CINAHL и Cochrane.
ВЫБОР ИССЛЕДОВАНИЙ: Исследования, в которых сообщается о пренатальной диагностике ОКС. Наблюдались следующие результаты: хромосомные аномалии при стандартном кариотипе и анализе хромосомного микрочипа (CMA), дополнительные аномалии, обнаруженные только на пренатальной МРТ и при постнатальной визуализации или клинической оценке, соответствие между пренатальной и послеродовой диагностикой и исходом нервного развития.
ИЗВЛЕЧЕНИЕ ДАННЫХ: Для объединения данных использовался мета-анализ пропорций.
РЕЗУЛЬТАТЫ: Было включено 27 исследований.В cACC хромосомные аномалии встречались в 4,81% (95% доверительный интервал [ДИ] 2,2–8,4). Общий и мелкий моторный контроль были ненормальными в 4,40% (95% ДИ, 0,6–11,3) и 10,98% (95% ДИ, 4,1–20,6) случаев, соответственно, тогда как в 6,80% (95% ДИ, 1,7–14,9) были представлены с эпилепсией. Аномальный когнитивный статус наблюдался в 15,16% (95% ДИ, 6,9–25,9) случаев. При частичном АКК частота хромосомных аномалий составляла 7,45% (95% ДИ 2,0–15,9). Контроль мелкой моторики нарушен в 11,74% (95% ДИ 0,9–32.1) случаев, и 16,11% (95% ДИ, 2,5–38,2) имели эпилепсию. Когнитивный статус был нарушен в 17,25% (95% ДИ, 3,0–39,7) случаев.
ОГРАНИЧЕНИЯ: Различные инструменты развития нервной системы и время наблюдения за включенными исследованиями.
ВЫВОДЫ: У детей с пренатальным диагнозом изолированного ОКК обнаруживается несколько степеней нарушения моторного контроля, координации, речи и когнитивного статуса. Однако ввиду большой разнородности показателей исходов, времени наблюдения и используемых инструментов нейроразвития необходимы большие проспективные исследования для установления фактического возникновения нейропсихологической заболеваемости у детей с изолированной ОЦК.
Однако ввиду большой разнородности показателей исходов, времени наблюдения и используемых инструментов нейроразвития необходимы большие проспективные исследования для установления фактического возникновения нейропсихологической заболеваемости у детей с изолированной ОЦК.
- ACC —
- агенезия мозолистого тела
- cACC —
- полная агенезия мозолистого тела
- CI —
- доверительный интервал
- CMA —
- хромосомный хром
- CN нервная система
- CNV —
- Вариация количества копий
- NOS —
- Шкала Ньюкасла-Оттавы
- pACC —
- Частичная агенезия мозолистого тела
Агенезия мозолистого тела является одной из наиболее ACC общие врожденные аномалии головного мозга с оценочной распространенностью от 1.От 8 на 10 000 в общей популяции до 230–600 на 10 000 у детей с нарушениями развития нервной системы. 1 — 3
Исходы нервного развития для лиц с каллозальными аномалиями чрезвычайно различаются даже у детей с одинаковыми нейроанатомическими профилями, и часто наблюдается значительное совпадение нейропсихологических показателей между пациентами с полным ОКК (сАСС) и пациентами с частичный ACC (pACC). 4 Задержка двигательных и когнитивных функций, эпилепсия, а также социальные и языковые дефициты являются наиболее частыми симптомами, о которых сообщается у лиц с ОКК; кроме того, ACC связывают с возникновением аутизма, шизофрении и нарушений внимания. 5 — 9 Однако педиатрические исследования смещены из-за того, что сообщаются только симптоматические случаи.
Достижения в методах пренатальной визуализации привели к увеличению частоты обнаружения ОКС; однако дородовое консультирование, когда у плода диагностирована эта аномалия, по-прежнему является сложной задачей. 5
Хромосомные аномалии распространены при ОКК, особенно когда присутствуют связанные аномалии, и пренатальные инвазивные тесты обычно проводятся во время беременности, чтобы исключить анеуплоидии.Хромосомный микрочип (CMA) позволяет обнаруживать небольшие делеции и дупликации генома, которые обычно не наблюдаются при стандартном цитогенетическом анализе (вариации числа копий [CNV]). Было показано, что плоды с аномалиями центральной нервной системы (ЦНС) и нормальным кариотипом имеют значительно более высокий риск генетических аномалий при анализе CMA; однако риск клинически значимых CNVs у плодов с изолированными каллозными аномалиями еще полностью не установлен. 10 , 11
Антенатальная МРТ обычно выполняется для исключения сопутствующих аномалий, которые являются основными определяющими факторами исхода в случаях ОКС; однако фактическая диагностическая точность МРТ плода при изолированном ОКК все еще обсуждается. 12
Исходы нервного развития у плодов с изолированными ОЦК в большинстве случаев считаются нормальными, особенно при полной агенезии. Однако для более правильного консультирования родителей требуется точная категоризация бремени нейропсихологических нарушений. 13
Первой целью этого систематического обзора было установить частоту ассоциированных генетических или анатомических аномалий у тех пациентов, у которых при первоначальном ультразвуковом обследовании были обнаружены изолированные ОКК; второстепенной целью было изучить статус развития нервной системы этих детей.
Методы
Протокол, критерии отбора, источники информации и поиск
Этот обзор был выполнен в соответствии с заранее разработанным протоколом и рекомендован для систематических обзоров и метаанализа. 14 , 15 В базах данных Medline, Embase, CINAHL и Cochrane 15 февраля 2014 г. был проведен электронный поиск с использованием комбинаций терминов соответствующих медицинских предметных заголовков, ключевых слов и вариантов слов для выражения «агенезия мозолистого тела» и «результат»; затем поиск был обновлен 26 ноября 2015 г. (Дополнительная таблица 5).Критерии поиска и отбора были ограничены английским языком. Списки ссылок на соответствующие статьи и обзоры были просмотрены вручную на предмет дополнительных отчетов. Руководства PRISMA были соблюдены. 16
Выбор исследований, сбор данных и элементы данных
Исследования оценивались по следующим критериям: популяция, тип агенеза мозолистой оболочки (cACC и pACC), тип оценки визуализации и исход (таблица 1).
ТАБЛИЦА 1Общая характеристика включенных исследований
Два автора (F.Д. и Г.П.) рассмотрели все тезисы независимо. Соглашение относительно потенциальной актуальности было достигнуто консенсусом; Были получены полнотекстовые копии этих статей, и те же два рецензента независимо друг от друга извлекли соответствующие данные, касающиеся характеристик исследования и исхода беременности. Несоответствия были обсуждены рецензентами, и был достигнут консенсус с третьим автором. Если для одной и той же когорты с идентичными конечными точками было опубликовано> 1 исследования, включали отчет, содержащий наиболее полную информацию о популяции, чтобы избежать дублирования популяций.Для тех статей, в которых информация не была представлена, но методология была такова, что эта информация должна была быть записана изначально, связывались с авторами.
Оценка качества включенных исследований проводилась с использованием шкалы Ньюкасла-Оттавы (NOS) для когортных исследований (таблица 2). Согласно NOS, каждое исследование оценивается по трем основным направлениям: выбор исследуемых групп, сопоставимость групп и интересующий результат установления. 44 Оценка выбора исследования включает оценку репрезентативности облученной когорты, выбор необлученной когорты, установление воздействия и демонстрацию того, что интересующий результат отсутствовал в начале исследования.Оценка сопоставимости исследования включает оценку сопоставимости когорт на основе дизайна или анализа. Наконец, установление интересующего результата включает оценку типа оценки интересующего результата, продолжительности и адекватности последующего наблюдения. Согласно NOS, исследованию можно присвоить максимум 1 звезду за каждый пронумерованный элемент в категориях «Выбор» и «Результат». За категорию сопоставимости можно присвоить максимум 2 звезды. 44
ТАБЛИЦА 2Оценка качества включенных исследований
Риск систематической ошибки, сводные измерения и обобщение результатов
Частота следующих исходов была проанализирована у плодов с пренатальной диагностикой cACC и pACC отдельно:
Хромосомные аномалии, обнаруженные с помощью стандартного анализа кариотипа.
Патогенные CNV в CMA.
Частота дополнительных аномалий ЦНС, обнаруженных только на пренатальной МРТ, но пропущенных при первоначальном сканировании.
Дополнительные аномалии ЦНС и вне ЦНС обнаруживаются только при послеродовой визуализации или клинической оценке, но не обнаруживаются при пренатальной визуализации.
Соответствие пренатальной и послеродовой диагностики.
Исход нервного развития.
В исследование были включены только плоды с пренатальным диагнозом ОКС с помощью трансабдоминального или трансвагинального ультразвукового исследования. cACC определяли как полное отсутствие всех анатомически определенных областей мозолистого тела, тогда как pACC определяли как наличие по крайней мере 1 области мозолистого тела.Для оценки частоты аномального кариотипа в анализ были включены только случаи изолированного ОЦК, определенные как не имеющие дополнительных аномалий ЦНС и вне ЦНС, обнаруженных при ультразвуковом сканировании. Были включены только те случаи, у которых полный кариотип был проверен пренатально или постнатально. По возникновению генетических аномалий, обнаруженных только при ЦМА, подходящими для анализа считались только плоды с изолированным АСС и нормальным стандартным кариотипом. Наличие дополнительных аномалий, выявляемых только на пренатальной и послеродовой МРТ, оценивали только у плодов без дополнительных аномалий и нормального кариотипа.Для целей этого исследования вентрикуломегалия от легкой до умеренной (определяемая как ширина бокового желудочка ≤15 мм) не была включена в качестве ассоциированной церебральной мальформации, поскольку ее развитие связано с реорганизацией мозга из-за агенеза мозолистой оболочки.
Исход развития нервной системы у младенцев с АКК был установлен исключительно в случаях изолированного ППК с нормальным полным стандартным кариотипом и без постнатально подтвержденных других аномалий ВНС и вне ЦНС. Случаи с изолированным ОЦК, подтвержденным на постнатальной визуализации, но показывающими экстрацеребральные аномалии при клиническом обследовании, в анализ не включались.Кроме того, поскольку в подавляющем большинстве исследований, показывающих влияние CMA у плодов с изолированным ACC, не сообщалось об исходе нервного развития, было невозможно провести субанализ, чтобы установить неврологический профиль тех случаев с нормальным стандартным кариотипом и клинически значимым. CNV найдены в CMA.
Исходы нервного развития были разделены на 3 разные категории (нормальные, пограничные / умеренные и тяжелые), как определено в исходном исследовании. Кроме того, чтобы обеспечить более объективную оценку неврологического статуса этих детей, мы также оценили исходы нервного развития с точки зрения: (1) общего моторного контроля, (2) мелкой моторики, (3) когнитивного статуса, (4) эпилепсии. , (5) визуальный контроль, (6) сенсорный статус, (7) язык и (8) координация.Все эти цифры были получены для плодов с cACC и pACC отдельно.
Только исследования, в которых сообщалось о пренатальном диагнозе ОКС, были сочтены подходящими для включения в текущий систематический обзор; послеродовые исследования или исследования, из которых не удалось выделить случаи, диагностированные пренатально, были исключены. Случаи с дисгенезией и / или гипоплазией мозолистого тела, а также случаи с отсутствием четкого определения аномалии не считались подходящими для включения. Исследования на основе аутопсии были исключены на том основании, что у плодов, подвергающихся прерыванию беременности, с большей вероятностью будут обнаружены связанные основные структурные и хромосомные аномалии.Исследования, сообщавшие о согласованности пренатальной и послеродовой диагностики ОКС, были исключены, если они не предоставили информацию о том, была ли аномалия изолированной или нет. Исследования неизолированных случаев ОКК были исключены, как и исследования, опубликованные до 2000 года, потому что мы чувствовали, что достижения в методах пренатальной визуализации и улучшения в диагностике и определении аномалий ЦНС делают эти исследования менее актуальными. Наконец, исследования, которые не давали четкой классификации аномалии, и исследования, в которых не проводилось различий между cACC и pACC, не были сочтены подходящими для включения в текущий обзор.Однако, поскольку было невозможно экстраполировать цифры встречаемости патогенных CNV у плодов с cACC и pACC отдельно, этот результат был установлен в общей популяции плодов с агенезом мозолистой оболочки.
Допускались только полнотекстовые статьи; Отчеты о случаях, тезисы конференций и серии случаев с <3 случаями ОКС, независимо от того, были ли аномалии изолированными или нет, также были исключены, чтобы избежать предвзятости публикации.
Мы использовали метаанализ пропорций для объединения данных. 45 Графики-воронки (дополнительные рисунки 10, 11, 12, 13 и 14), отображающие частоту результатов отдельных исследований в сравнении с их точностью (1 на SE), были выполнены с исследовательской целью. Тесты на асимметрию графика воронки не использовались, когда общее количество публикаций, включенных для каждого результата, было <10. В этом случае мощность тестов слишком мала, чтобы отличить случайность от реальной асимметрии. 45 , 46
Гетерогенность между исследованиями была исследована с использованием статистики I 2 , которая представляет собой процент вариации между исследованиями, которая обусловлена гетерогенностью, а не случайностью.Значение 0% указывает на отсутствие наблюдаемой неоднородности, тогда как значения I 2 ≥50% указывают на существенный уровень неоднородности. Модель с фиксированными эффектами использовалась, если не было значительной статистической неоднородности. Напротив, если были доказательства значительной неоднородности между включенными исследованиями, использовалась модель случайного эффекта. 47
Все мета-анализы пропорций были выполнены с использованием StatsDirect версии 2.7.9 (StatsDirect, Ltd, Altrincham, Чешир, Соединенное Королевство).
Результаты
Выбор исследований и характеристики
Всего было идентифицировано 2296 статей, 153 были оценены на предмет их соответствия критериям включения (дополнительная таблица 6), а 27 исследований были включены в систематический обзор (рис. 1). 17 xref ref-type = «bibr» rid = «B18» /> — 43 Эти 27 исследований включали 484 плода с изолированным АЦП и отсутствием других связанных аномалий ЦНС и / или экстра-ЦНС при первой пренатальной оценке.
РИСУНОК 1Блок-схема систематического обзора.
Оценка качества включенных исследований проводилась с использованием БДУ для когортных исследований. 44 Некоторые из включенных в обзор исследований показали в целом хорошие результаты в отношении выбора и сопоставимости исследуемых групп и установления интересующего результата. Основные недостатки этих исследований заключались в их ретроспективном дизайне, небольшом размере выборки и отсутствии стандартизированного послеродового подтверждения.Кроме того, относительно короткий период наблюдения после родов не позволил точно оценить общую частоту дополнительных аномалий, обнаруженных только после рождения и пропущенных пренатально.
Обобщение результатов
cACC
В этот систематический обзор были включены 20 исследований, включающих 261 плод с изолированным cACC.
Частота хромосомных аномалий составила 4,81% (95% доверительный интервал [ДИ], 2,2–8,4) (рис. 2, таблица 3). Цифры для различных хромосомных аномалий, обнаруженных у плодов с изолированным сАСС, показаны в дополнительной таблице 7.
РИСУНОК 2Объединенные доли встречаемости хромосомных аномалий и патогенных CNV у плодов с cACC и pACC.
ТАБЛИЦА 3Объединенные пропорции исходов, изученных в этом систематическом обзоре у плодов с cACC
Не удалось экстраполировать данные о частоте клинически значимых CNV у плодов с изолированным cACC и нормальным кариотипом, что привело к появлению клинически значимых CNV. оценивали у плодов с помощью cACC или pACC.
В целом, частота значимых CNV у плодов с изолированным ACC (cACC или pACC) и нормальным кариотипом составила 5.74% (95% ДИ, 1,3–13,1) (рис. 2).
В 2,99% (95% ДИ, 0,9–6,1) случаев пренатальная диагностика не позволила правильно определить cACC, а в некоторых случаях pACC ошибочно диагностировали как имеющую cACC (дополнительный рисунок 5).
Дополнительные аномалии, не обнаруженные при пренатальном УЗИ, были диагностированы при МРТ плода в 7,83% (95% ДИ, 1,2–19,6) случаев, тогда как частота дополнительных структурных аномалий, диагностированных только после рождения и пропущенных при пренатальной оценке, составила 5,49% ( 95% ДИ 2,4–9.7) (Таблица 3, дополнительные рисунки 6 и 7). Описание отдельных случаев аномалий, обнаруженных только при МРТ плода и постнатальной визуализации / клиническом исследовании, показано в дополнительных таблицах 8 и 9.
Ввиду высокой неоднородности дизайна исследования, возраста и типа оценки, а также времени последующего наблюдения: выше, показатели аномальных исходов развития нервной системы могут не отражать фактические нейропсихологические характеристики этих детей и должны интерпретироваться с осторожностью. Кроме того, было невозможно установить показатели развития нервной системы у детей с нормальным стандартным полным кариотипом и без CNV на CMA, потому что только одно исследование сообщило об этом результате.Исходы нервного развития были нормальными у 76,04% (95% ДИ, 64,3–86,1) детей с пренатальным диагнозом изолированной САСС, подтвержденным при рождении (рис. 3, табл. 4). Частота пограничных / умеренных и тяжелых исходов нервного развития у этих детей составляла 16,04% (95% ДИ, 7,6–26,8) и 8,15% (95% ДИ, 2,5–16,8) соответственно. В таблице 3 представлены подробные цифры аномальных показателей развития нервной системы у детей с изолированной САСС. У 4 пациентов был нарушен контроль крупной и мелкой моторики.40% (95% ДИ 0,6–11,3) и 10,98% (95% ДИ 4,1–20,6) случаев, тогда как 6,80% (95% ДИ 1,7–14,9) этих детей имели эпилепсию. Когнитивный статус был затронут в 15,16% (95% ДИ, 6,9–25,9) случаев, тогда как нарушение речи было затронуто в 8,02% (95% ДИ, 2,1–17,3). Наконец, аномальный глазной контроль и координация наблюдались в 15,84% (95% ДИ, 4,3–32,9) и 9,50% (95% ДИ, 3,2–18,7) случаев соответственно (дополнительный рисунок 8).
РИСУНОК 3Объединенные пропорции возникновения аномальных исходов развития нервной системы у плодов с САСС.
ТАБЛИЦА 4Объединенные пропорции исходов, изученных в этом систематическом обзоре у плодов с pACC
Индивидуальные описания исходов у детей с изолированным cACC, показывающим аномальные профили нервного развития, показаны в дополнительной таблице 10.
pACC
Пятнадцать исследований, включающих 225 плодов с pACC были включены в этот обзор.
Частота хромосомных аномалий у плодов с pACC и отсутствием других структурных аномалий, видимых при пренатальной визуализации, составляла 7.45% (95% ДИ, 2,0–15,9) (рис. 2, таблица 4). Цифры для различных хромосомных аномалий, обнаруженных у плодов с изолированной pACC, показаны в дополнительной таблице 11.
Дополнительные аномалии, не обнаруженные при пренатальном УЗИ, были диагностированы на МРТ плода в 11,86% (95% ДИ, 3,2–24,9) случаев, тогда как частота дополнительных структурных аномалий, диагностированных только после рождения и пропущенных при пренатальной оценке, составила 14,46% (95% ДИ, 6,7–24,6) (таблица 4, дополнительные рисунки 6 и 7). Описание отдельных случаев аномалий, обнаруженных только при МРТ плода и послеродовой визуализации / клиническом исследовании, показано в дополнительных таблицах 12 и 13.
Несоответствие между пренатальным и постнатальным диагнозом pACC произошло в 7,99% (95% ДИ, 2,5–16,3) случаев, в основном состоящих в случаях гипопластического или дисгенетического мозолистого тела, ошибочно диагностированного как pACC (дополнительный рисунок 5).
Оценка исходов развития нервной системы у детей с изолированной pACC была еще более проблематичной из-за меньшего размера анализируемой выборки по сравнению с cACC.
Исходы нервного развития были нормальными в 71,42% (95% ДИ, 53.1–86.7) детей с пренатальным диагнозом изолированной ПКК, подтвержденным при рождении (таблица 4). Частота пограничных / умеренных и тяжелых исходов нервного развития у этих детей составила 14,92% (95% ДИ, 4,2–30,7) и 12,52% (95% ДИ, 2,9–27,5), соответственно (рис. 4).
РИСУНОК 4Объединенные пропорции возникновения аномальных исходов развития нервной системы у плодов с pACC.
Контроль мелкой моторики был нарушен в 11,74 (95% ДИ, 0,9–32,1) случаев и в 16,11% (95% ДИ, 2,5–38.2) этих детей с эпилепсией. Когнитивный статус был затронут в 17,25% (95% ДИ, 3,0–39,7) случаев, тогда как нарушение речи было замечено в 17,25% (95% ДИ, 3,0–39,7) случаев. Наконец, нарушение координации происходило в 11,74% (95% ДИ, 0,9–32,1) случаев (дополнительный рисунок 9).
Индивидуальные описания исходов у детей с изолированной pACC, показывающие аномальный профиль нервного развития, показаны в дополнительной таблице 14.
Discussion
Summary of Evidence
Результаты этого систематического обзора показали, что у плодов с изолированной мозолистой агенезией (cACC или pACC) подвержены высокому риску хромосомных аномалий.Даже когда стандартное кариотипирование является нормальным, все равно существует значительный риск генетических аномалий, обнаруживаемых только при анализе CMA. В случаях пренатальной диагностики изолированной АКК риск ассоциированных аномалий, обнаруживаемых только на МРТ плода, составляет около 8% и 12% у плодов с цАСС и пАСС, соответственно, тогда как связанные аномалии, обнаруженные только после рождения, могут возникать примерно в 5% случаев. плоды с САСС и у 14% плодов с РАСС. Короткие периоды наблюдения, неоднородность протоколов визуализации, используемые инструменты развития нервной системы, расхождения в определении аномального исхода и небольшое количество включенных случаев не позволили нам сделать какие-либо надежные выводы относительно возникновения аномальных исходов нервного развития у детей с пренатальный диагноз изолированной мозолистой агенезии.Результаты этого систематического обзора показали, что около двух третей детей показали нормальный исход развития нервной системы, хотя у значительной части этих детей могут быть нарушены мелкая и грубая моторика, координация, язык и когнитивный статус. Однако эти цифры могут не отражать фактическое бремя нейропсихологической заболеваемости у детей с изолированным ОКК; Для подтверждения этих результатов необходимы дополнительные большие проспективные исследования.
Сильные стороны и ограничения
Сильные стороны этого исследования — его надежная методология для выявления всех возможных исследований, оценки качества данных и синтеза всех подходящих данных.
Для нескольких метаанализов количество включенных исследований было небольшим, а некоторые исследования включали небольшое количество. Оценка потенциальной систематической ошибки публикации также была проблематичной либо из-за характера результата (частота с левой стороной ограничена значением 0), что ограничивает надежность графиков воронки, либо из-за небольшого количества отдельных исследований, которые сильно ограничивает надежность формальных тестов. Кроме того, все включенные исследования были ретроспективными и, следовательно, подвержены значительному риску систематической ошибки отбора.Кроме того, во многих исследованиях адекватно не сообщалось о некоторых исходах и ассоциациях. Наконец, из-за относительно короткого постнатального периода наблюдения общая частота дополнительных аномалий, обнаруживаемых только после рождения и пропущенных пренатально, могла быть недооценена.
Оценка исходов развития нервной системы у детей с пренатальным диагнозом изолированной ОКК также была проблематичной; различия в возрасте на момент последующего наблюдения и используемые инструменты нейроразвития не позволили провести значимую стратификацию различных показателей исходов; следовательно, приведенные в текущем обзоре данные по отклонениям в развитии могут не отражать фактическое бремя нейропсихологических сопутствующих заболеваний, связанных с изолированным ОКК, и их следует интерпретировать с осторожностью.Кроме того, было невозможно стратифицировать анализ, включающий только плоды с нормальным стандартным полным кариотипом и без патогенных CNV, обнаруженных в CMA, ввиду отсутствия данных относительно исходов развития нервной системы в этих исследованиях. В этом сценарии вполне возможно, что в анализ были включены случаи с изолированным ACC, нормальным стандартным кариотипом и патогенными CNV, что исказило результаты. Наконец, в большинстве включенных исследований не сообщалось подробного описания неврологических характеристик плодов с изолированными ОЦК, а анализировался лишь стратифицированный анализ по 3 различным категориям (нормальный, пограничный / умеренный и тяжелый), для которых критерии включения различались между исследования.Ввиду всех этих ограничений к итоговым суммарным показателям следует относиться с некоторой осторожностью.
Несмотря на все эти ограничения, наш обзор представляет собой самую современную общую оценку исхода нервного развития при агенезе мозолистой оболочки, диагностированного пренатально; это важно, потому что консультирование родителей, основанное на отдельных небольших исследованиях, которые подвержены предвзятости публикации, может быть неадекватным.
Значение для клинической практики и перспективы на будущее
Достижения в методах пренатальной визуализации привели к повышению диагностической точности ультразвукового исследования при обнаружении аномалий мозолистой оболочки.Однако пренатальное консультирование, когда у плода диагностирован ОКК, является сложной задачей.
Результаты этого систематического обзора показали, что хромосомные аномалии могут возникать у значительной части плодов с изолированной АСС; более того, риск генетических аномалий, не обнаруженных с помощью обычного кариотипирования, также не является незначительным. Недавно было показано, что CMA предоставляет полезную информацию для пациентов с нарушениями обучаемости и врожденными аномалиями, для которых обычные цитогенетические тесты оказались отрицательными.Результаты этого обзора подтверждают использование CMA при пренатальной диагностике ACC. 48
МРТ плода обычно выполняется в случаях пренатальной диагностики ОКС. В текущем обзоре ассоциированные аномалии, не обнаруженные при УЗИ, были диагностированы в 7,83% (95% ДИ, 1,2–19,6) и в 11,86% (95% ДИ, 3,2–24,9) в cACC и pACC, соответственно. Однако даже в случаях пренатальной диагностики изолированной аномалии риск того, что АКК не будет полностью изолирован, относительно высок, с дополнительными аномалиями, обнаруженными только при послеродовой визуализации и / или клиническом обследовании, но пропущенными пренатально, что встречается в 5 случаях.49% (95% доверительный интервал [ДИ], 2,4–9,7) и 14,46% (95% доверительный интервал [ДИ], 6,7–24,6) плодов с pACC и cACC, соответственно.
Количественная оценка реального вклада МРТ плода в аномалии головного мозга является сложной задачей. Несколько факторов, таких как опыт оператора, протокол визуализации, время и тип оценки, интервал между УЗИ и МРТ, а также тип аномалии, могут играть роль в этом сценарии и объяснять широкую неоднородность и противоречивые результаты, о которых сообщалось в ранее опубликованных исследованиях.Несмотря на все эти противоречия, МРТ обычно используется в клинической практике для подтверждения диагноза и поиска связанных аномалий. Подавляющее большинство дополнительных аномалий, обнаруживаемых только при МРТ плода, связаны с нарушениями миграции нейронов (дополнительные таблицы 8 и 12), которые могут быть обнаружены преимущественно в третьем триместре беременности. Исходя из этого, когда МРТ выполняется во время сканирования аномалий для подтверждения диагноза, было бы разумно организовать последующее сканирование в третьем триместре, чтобы установить, действительно ли изолирован АКК.Эти предложения основаны на опыте авторов, и для подтверждения этих результатов необходимы дальнейшие исследования оптимального времени проведения МРТ плода.
Более того, даже когда пренатальная диагностика исключает связанные аномалии, все еще существует значительный риск (5,5% и 14,5% у плодов с cACC и pACC, соответственно) для обнаружения дополнительных аномалий после рождения (дополнительные таблицы 8 и 12). Это следует подчеркнуть во время дородового консультирования, поскольку пренатальная визуализация не всегда позволяет различить сложные и изолированные случаи, и что послеродовая визуализация и тщательное клиническое обследование необходимы для подтверждения того, что ОКК действительно изолирован.
Оценка профиля нервного развития у детей с ОКК является сложной задачей. Термин «исход нервного развития» может вводить в заблуждение и не подходить при работе с аномалиями головного мозга, поскольку он охватывает широкий спектр признаков с различными лежащими в основе расстройствами и патологическими процессами, которые не всегда легко измерить и которые представляют собой непрерывное взаимодействие между патологическими, средовыми и адаптивными факторами. Сообщается, что интеллектуальные способности людей с ОКК находятся в нижнем диапазоне нормы; кроме того, у значительной части детей нарушены или нарушены трудности в практических языковых и математических навыках, экспрессивной и восприимчивой речи, визуальном и пространственном мышлении и навыках внимания. 5 Однако постнатальные исследования смещены из-за того, что включены только пациенты с симптомами, что потенциально переоценивает бремя инвалидности, наблюдаемое при этих аномалиях.
Результаты этого систематического обзора подтвердили эти результаты и показали, что у детей с ОКК могут быть разные степени нарушений в неврологической и нейропсихологической областях.
Хотя прямое сравнение нервно-психических показателей у детей с САСС и детей с РАСС не проводилось ввиду дизайна большинства включенных исследований, которые не позволяли проводить такое сравнение, результаты этого обзора действительно не предполагают огромной разницы между двумя различными субъектами агенеза мозолистой оболочки.Результаты этого метаанализа удивительны и не согласуются с тем, что наблюдается после рождения, когда pACC с меньшей вероятностью будет диагностирован как отдельный результат и обычно зависит от более высоких показателей нарушений развития нервной системы по сравнению с cACC. По мнению коллективных авторов, относительно высокая частота благоприятных исходов, наблюдаемых при pACC, может быть связана с тем, что во многих случаях, отмеченных пренатально как pACC, после рождения диагностируется гипоплазия мозолистого тела.
Выводы
Плоды с изолированной каллозальной агенезией имеют высокий риск хромосомных аномалий, даже если стандартный кариотип отрицательный. Пренатальная визуализация не может полностью исключить связанные аномалии, обычно сосуществующие с этим состоянием, и риск того, что АКК не будет полностью изолирован после рождения, является значительным.
При изолированной агенезии мозолистой оболочки у значительной части детей могут возникать аномалии мелкой и крупной моторики, координации, языка, когнитивного статуса и интеллекта.Однако ввиду небольшого числа включенных случаев, короткого периода наблюдения и неоднородности принятых инструментов нейроразвития, эти результаты следует интерпретировать с осторожностью, и в будущих крупных проспективных исследованиях, направленных на оценку нервно-психического развития и психологических показателей детей с Изолированная мозолистая агенезия с использованием стандартизованных инструментов оценки нервного развития через соответствующие промежутки времени необходима для установления фактических нейропсихологических показателей и интеллектуальных нарушений у детей с изолированными ОКК.
Благодарности
Мы благодарим профессора О. Гленна, профессора Дж. Барковича, профессора Л. Читти, профессора Г. Касприана, профессора Д. Прайера, профессора Берга, профессора В. Д’Аддарио, профессора П. Вольпе, профессора Г. Риццо, профессор М. Килби, доктор А. Руланд, профессор Т. А. Хьюисман, доктор О. Шен, профессор В. Браун, доктор А. Языджиоглу, профессор П. Х. Тан, доктор Ф. Хадзагич-Катибусич, доктор Х. Слейтер, доктор М. Ямасаки , Д-р Ф. Скотт, д-р Ю. Йинон, д-р М. Нанни, д-р А. Кнафель, д-р Л. Климан, д-р С. Пашай, д-р Ф. Скотт, д-р Э.М. Вестергаард, д-р Г. Сребняк, д-р С. Тору и д-ру I Papoulidis за их вклад в этот систематический обзор с точки зрения предоставленных дополнительных данных и поддержки.
Сноски
- Адресная корреспонденция Франческо Д’Антонио, MD, PhD, Департамент клинической медицины, Факультет медицинских наук, UiT — Арктический университет Норвегии, Hansine Hansens veg 18, 9019 Tromsø, Norway. Электронная почта: francesco.dantonio {at} uit.no
ФИНАНСОВАЯ ИНФОРМАЦИЯ: Авторы указали, что у них нет финансовых отношений, имеющих отношение к этой статье, которые следует раскрывать.
ФИНАНСИРОВАНИЕ: Нет внешнего финансирования.
ПОТЕНЦИАЛЬНЫЙ КОНФЛИКТ ИНТЕРЕСОВ: Авторы указали, что у них нет потенциальных конфликтов интересов, которые следует раскрывать.